Research Article |
Open Access |
|
|
Vectors and Integration in Gene
Therapy: Statistical Considerations |
Alessandro Ambrosi 1, Clelia Di Serio 1* |
1University Centre of Statistics for Biomedical Sciences (CUSSB),
Università Vita-Salute San Raffaele, Via Olgettina, 58 20132 Milano |
| *Corresponding author: |
Dr. Clelia Di Serio, University Centre of Statistics for Biomedical Sciences (CUSSB),
Università Vita-Salute San Raffaele, Via Olgettina, 58 20132 Milano,
Email: diserio.clelia@hsr.it |
|
| Received February 23, 2009; Accepted February 25, 2009; Published February 27, 2009 |
|
Citation: Alessandro A, Di Serio C (2009) Vectors and Integration in Gene Therapy: Statistical Considerations. J Comput Sci
Syst Biol 2: 117-123. |
| |
Copyright: © 2009 Alessandro A, et al. This is an open-access article distributed under the terms of the Creative Commons
Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author
and source are credited. |
| |
|
In gene therapy the integration process of the viral DNA genome into the host cell genome is a necessary step
for virus integration. Just few years ago, retrovirus integration was believed to be random and the chance of
accidentally activating a gene was considered remote. It has been seen that this process is not random and
different viruses may show different preferences to integrate in some specific areas of the genome. Tumorigenesis
associated to some studies in gene therapy is suspected to be caused by insertion process. Depending on
whether the provirus integrates into or in the vicinity of genes (Transcription Start Sites, TSS), normal trascription
can be enhanced or disrupted thus inducing oncogenic mutations. This is called insertional mutagenesis.
Investigating whether an area over the genome could be favoured by retrovirus integration is a crucial aspect
in gene therapy. These area are called “Common Integration Sites”(CIS)or "hotspots". In the paper we stressed the importance of developing statistical procedures leading to a unique definition of CIS rather than a problem
related definition. We here propose some statistical solutions for the search of hotspots based on the Peaksheight
distribution, which account within the null hypothesis for the possible non-random behaviour of the integrations. |
Background |
| Gene therapy is a form of molecular medicine which treats
genetic diseases by replacing a defective gene, responsible
for the pathology, with a functional one. The basic principle
is to introduce a piece of genetic material into cells via a
virus which represents the vector for gene therapy. The
virus integrates with the cell DNA and thus delivers the
genetic material into the cell nucleus. This process is called
integration and may alter the host cells DNA. Recent studies
based on cellular and animal models (Bushman:2005)
reported empirical evidence of preference for certain
retroviral vectors, i.e. those deriving from Moloney Murine
Leukemia Virus (MLV), to integrate near the start of transcriptional
units, whereas others (like Simian Immunodeficiency Virus (SIV)- and Human Immunodeficiency Virus
(HIV)-based vectors) did not show the same tendency. The
mutation may alter the expression of genes in the vicinity of
the insertion or, when inserted within a gene, alter the gene
product. When the affected gene is a cancer gene (either a
proto-oncogene or a tumor suppressor gene), activation of
the proto-oncogene or inactivation of the tumor-suppressor
gene can cause uncontrolled proliferation (cell division) of
cells. Eventually this may give rise to tumors. These cancer-
causing insertions are referred to as insertional mutagenesis
or oncogenic integration. A tumor could develop
when an accumulation of oncogenic insertions causes uncontrolled
proliferation of a cell. This has been seen both in animal as well as human models. The related problem of
safety of a vector is a major hurdle (Montini et al. 2006). It
has been observed that in retroviral integration different
vectors show distinct target site preferences, thus finding a
unique statistical criteria to detect accumulation of integration
is a fundamental tool within the debate on safety of a
vector. (Recchia et al., 2006, Cassani et al., 2006). Some
approaches provided statistical and mathematical modelling
to to test the hypothesis of randomness (Abel et al 2007,
Ambrosi et al 2008). Moreover in the recent literature
(Cattoglio et al 2007) it has been proved that analysis of
MLV integration patterns in natural or experimentally induced
leukemias/lymphomas showed the existence of insertion
sites recurrently associated with a malignant phenotype.
These common insertion sites (CIS), also called
hotspots which include proto-oncogenes or other genes
associated with cell growth and proliferation, may present
when activated a causal relationship with the establishment
and/or progression of cancer. The definition of hotspot is however
not unique and crucially problem related”. A first model
to define a hotspot has been provided by Suzuki et al. (2002) and compares the mapped locations of the proviruses in the isolated tumors to randomly generated integrations from
100,000 Monte Carlo trials.This was done to determine cutoffs
for defining when two or more integrations in close
proximity were significant enough to assume that it didnt
happen by chance. Basically the cutoffs were within 30 kb
for 2 integrations, 50 kb for 3 insertions or 100 kb for 4
integrations. In terms of the null hypothe sis for hotspot analysis this is
problematic. Definition of hotspots is based on a comparison
to a random set, but there is a clear preference in integration
that should be taken into account in the null hypothesis. |
Wu et al. (2005) showed that pre-established role in cancer
is not sufficient support for the efficacy of the Suzuki
CIS technique. For instance expression level in MLV inte
gration may also play a role since MLV integrations are
biased towards genes with higher expression levels. A first
interesting way to account for this non-randomness in the
null hypothesis is the Wu et al. model which all ows 75% of
the integrations to occur randomly and 25% to integrate in a
Poisson distribution T5 kb around the transcriptional start
site. |
This introduction was aimed at highlighting how statistics
and probability must play a fundamental role in establishing
a criteria to detect accumulation sites and preferences
of integration for a better understanding of how safe
is a vector in gene therapy. To do this we address the following
question: how can we distinguish the preferences of
viruses to integrate close to TSS from their accumulation
due to some other reason (for instance to the presence of
some particular gene)?. |
Data and Experimental Design |
| In this paper we analyzed data derived from retroviral
transduction in T cells from leukemic patients treated with
allogeneic stem cell transplantation and donor lymphocytes
genetically modified with a suicide gene (HSV-TK).
Retroviral vectors integrate preferentially within or near transcribed
regions of the genome, with a preference for sequences
around promoters and for genes active in T cells at
the time of transduction. For details on the whole data set see Ambrosi et al. (2008). The following information are reported: |
- nucleotide (integration position)
- chromosomes
- integration distance from the TSS of genes in a window of 100 kb
- expression data for genes involved in the integration (hotspots and all)
- gene density in 1Mb neighbourhood
|
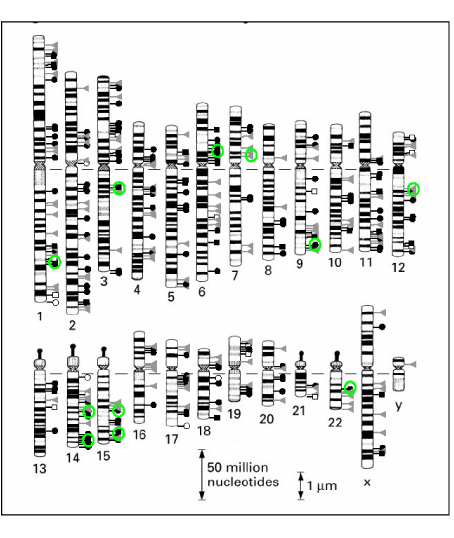
Figure 1 provides an example of Common Integration Site
(CIS) which are classified on the criteria of two integrations
occurring in a 100kb window. These can be visualized
on all chromosomes by high concentration of hit in a small
genomic area. |
|
Figure1: Integration site analysis in T cells. Green circles denote high integration density areas.
|
|
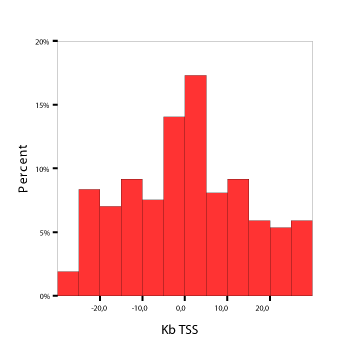
The observed distribution of integration distances from
TSS of the nearest gene are provided in Figure 2. This distribution
seems consistent with the new findings in
gene therapy literature (among other Ambrosi et al. 2008)
that TSS sort of attracts integrations. |
|
Figure 2: Integration site analysis in T cells. Green circles denote high integration density areas.
|
|
In this paper we provide some statistical proposals to investigate
the real distributional nature of a hotspot. As
mentioned above we want to address the following question:
does the integrations distribution reflects a virus
natural attitude to integrate in some genetic areas (like
TSS) or are rather some genetic areas that do attract integrations due to their functional characteristics? |
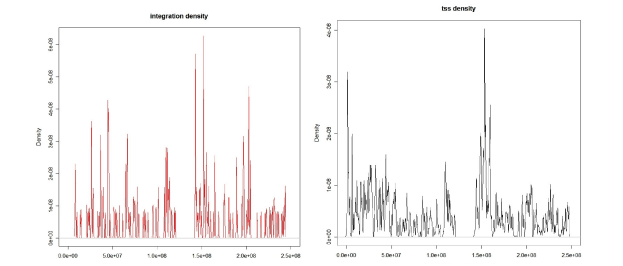
To explore this idea we compare the integration density
distribution with the TSS density distribution (which reflects
the gene density) like in Figure 3 (referred to the first chromosome).
Since the focus of this paper is on the statistical
procedure we illustrate our analysis with a small sample of
integrations on chromosome 1 only. |
|
Figure 3: Comparison between the integration density distribution and TSS density distribution.
|
|
It can easily be observed that the two distributions show
similarities. This is a natural consequence of the fact that
this virus integrates preferably close to the TSS and thus
more frequently in high gene density areas. We next focus
on those areas that attract insertions of the retrovirus even
when no high gene density is revealed. |
Statistical Procedure: the Peaks-Hight (P-H) Method |
| A natural way to provide a statistical approach for the
identification of CIS in distributional terms is a kernel estimation
procedure (Ridder et al. 2006) to find the regions in
the genome that show a significant increase in insertion
density. |
For any position over the genome, an estimate of the number
of insertions is obtained by summing all the kernel functions.
(rectangular , Barlett-Epanechnikov, etc.). |
Actually, the basic idea is to model non parametrically the
probability that an observation x will fall into a certain region,
that is F =∫ f ( x ) dx with F a smoothed (or averaged version of the density function f(x). The kernel density
estimator fb(x) for the estimation of the density value
f (x) at target point x is a local average smoother that, for
random variable xi in a prediction space calculate an average
of the observations in a neighbourhood of the target
point: |
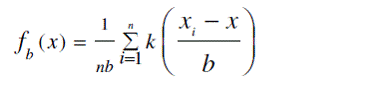 |
where k(×) denotes the Kernel function and b is the bandwidth
parameter which determines how large a
neighbourhood of the target point is. A large bandwidth generates
a smoother curve, while a small bandwidth generates
a wigglier curve, thus the choice of b being fundamental
and much more important than the choice of kernel
(Hastie and Tibshirani (1990)). We use here a standard
Gaussian kernel. |
Analys is is based on discrete data points indicating the
integration position. We are trying to establish how unusual
is the spatial patterning of these points. By turning the discrete
points into a continuous surface using Kernel estimation,
the data can then be explored. In particular we focus
on the maximum of the observed integration distribution estimated
with by means of a Gaussian kernel. This is now
our new random variable, Xi indicating integration peaks height
(P-H). |
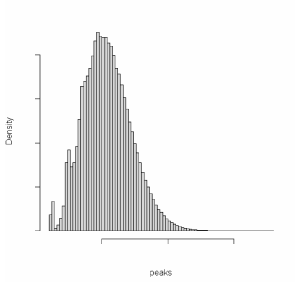
In the same estimation context we set the null hypothesis,
H0: integrations occur randomly over the genome (theoretical peaks distribution, Figure 4) to account for natural
integration preferences. To build this in terms of peaks
we compute the maximum over 1400 integrations which
are resampled from a uniform distribution 50000 times via
Monte Carlo method. |
|
Figure 4: Theoretical peaks distribution under the null hypothesis for Chromosome 1.
|
|
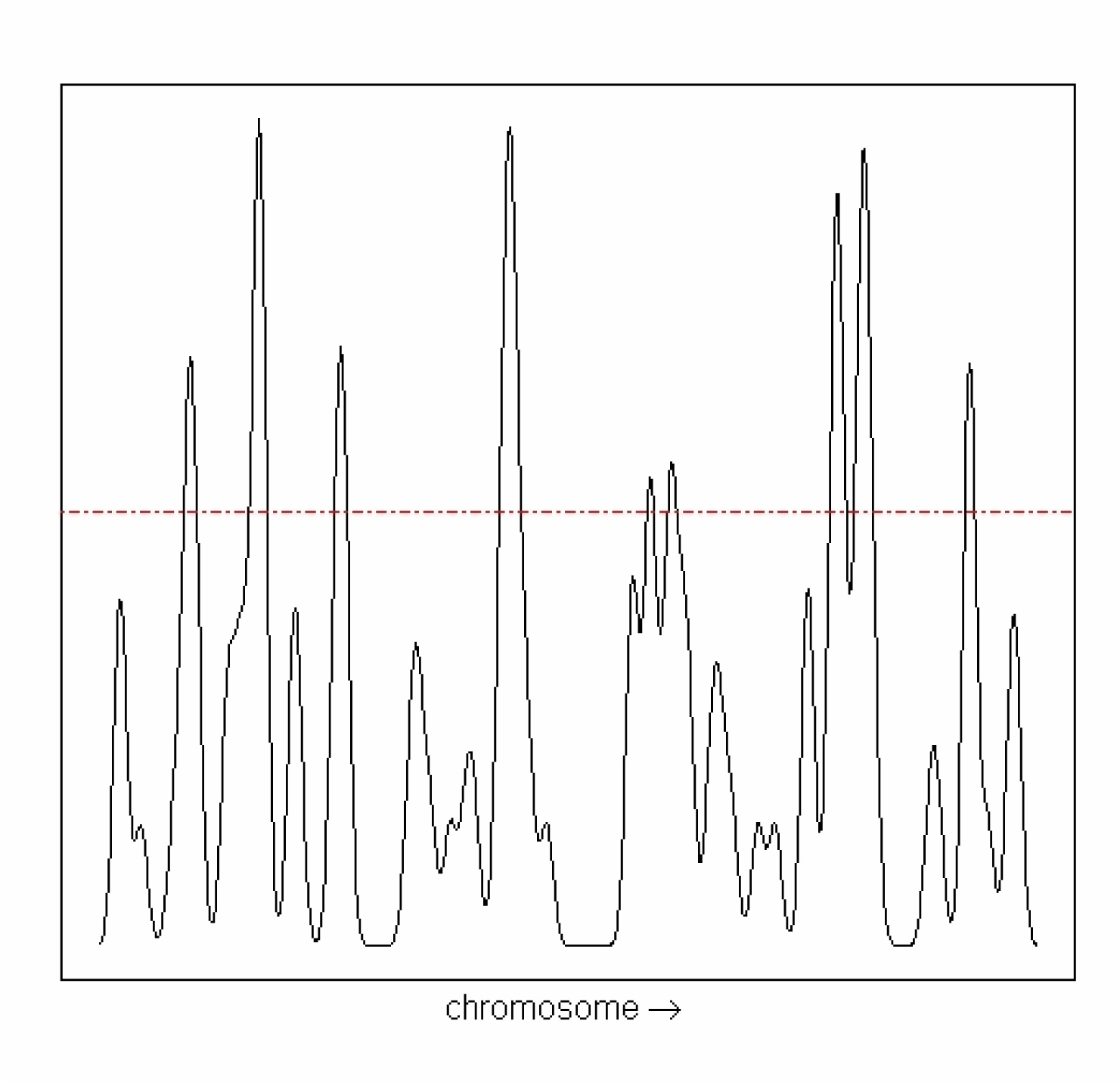
p-values are then computed for each estimated P-H value
from the theoretical peaks height distribution (Figure 5). |
|
Figure 5: Observed peaks-height distribution. Red line shows the significative peaks with alpha =0.05.
|
|
Correction for multiple comparisons is then applied, and
significant P-H values are extracted. The neighbourhood of
these identifies the P-H hotspot”. In this contribution we
present results on Chromosome 1 only. Results reported in
Table 1 lead to identify nucleotide positions where a real
hotspot occurs (that is based on P- based definition of
Hotspot). 7 hotspots can be identified after p-value corrections
for multiple testing. This is a first step that could deal to examine in terms of expression and properties the corresponding
genomic areas. |
Table 1: Estimation of the peaks and p-values corrections for multiple testing (Beniamini-Hochberg, Holm).
|
|
Final Remarks |
| The goal of this contribution was to provide some statistical
considerations on the real nature of a hotspot. Statistical
criteria for the identification of regions which are favoured
by integrations (CIS or hotspot) are needed. We approached this problem
by considering CIS not just like an area with very close
integrations but like an area with very high integration density.
Thus, we provide the null hypothesis based on kernel
estimation of the P-H distribution, when integration are uniformly
distributed over the genome. This criteria can be
extended by considering a rectangular kernel, which better
resemble the finite support of the integration distribution.
Moreover, based on the proposed criteria, we can compare the “PH hotspot” with the transcription start site distribution to distinguish which “P-H hotspot” reflects the high gene density
areas, and which can really be thought as a real h
otspot and thus leading to genetic investigations. |
Aknowledgements |
| We greatly aknowledge Barbara Cassani, Alessandro Aiuti and Fulvio Mavilio for helping in understanding the crucial aspects of data and biological issue. |
References |
- Ambrosi A, Cattoglio C, Di Serio C (2008) Retroviral
Integration Process in the Human Genome: Is It Really
Non-Random? A New Statistical Approach. PLoS
Comput Biology Vol. 4, Issue 8 / e1000144.
- Abel U, Deichmann A, Bartholomae C,
Schwarzawaelder K , Glimm H, et al. (2007) Real-time
definition of non-randomness in the distribution of genomic
events. PLoS ONE 2: e570. [ FIND THIS ARTICLE ONLINE ]
- Aiuti A, Ambrosi A, Di Serio C, Bordignon et al. (2007) Multilineage hematopoietic reconstitution without clonal
selection in ADA-SCID patients treated with stem cell
gene therapy. Journal of Clinical investigation in press
- Bushman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, et
al. (2005) Genomewide analysis of retroviral DNA integration.
Nat Rev Microbiol 3: 848-858. [ FIND THIS ARTICLE ONLINE ]
- Cassani B, Andolfi G, Di Serio C, Ambrosi A, et al. (2006)
Clonal analyses of retroviral vector integrations in ADASCID
patients treated with stem cell gene therapy.
Blood 108: 927A-927A 3249.
- Cattoglio C, Facchini G, Sartori D, Antonelli A, Miccio
A, et al. (2007) Hot spots of retroviral integration in human
CD34(+) hematopoietic cells. Blood 110: 1770-1778. [ FIND THIS ARTICLE ONLINE ]
- Hastie TJ, Tibshirani RJ (1990) Generalized Additive
Models, New York: Chapman and Hall.
- Hematti P, Hong B, Ferguson C, Adler R, Hanawa H, et
al. (2004) Distinct Genomic Integration of MLV and SIV
Vectors in Primate Hematopoietic Stem and Progenitor
Cells. PLoS Biol 2: 2183-2190.
- Jeroen R, Anthony U, Jaap K, Marcel R, Lodewyk W
(2006) Detecting Statistically Significant Common Insertion
Sites in Retroviral Insertional Mutagenesis
Screens. PLoS Computational Biology Volume 2, Issue
12.
- Rick S, Mitchell, Brett FB, Astrid RW, Schroder2, et al.
(2004) Retroviral DNA Integration: ASLV, HIV, and
MLV Show Distinct Target Site Preferences. PLoS Biology
Volume 2 | Issue 8 | e234.
- Montini E, Cesana D, Ambrosi A, Di Serio C, Naldini L,
et al. (2006) Hematopoietic stem cell gene transfer in a
tumor prone mouse model uncovers low genotoxicity of
lentiviral vector integration. Nature Biotec. 24: 687-96.
- Recchia A, Mavilio F, et al. (2006) Retroviral vector integration
deregulates gene expression but has no consequence
on the biology and function of transplanted T
cells. PNAS 103: 14571462. [ FIND THIS ARTICLE ONLINE ]
- Suzuki T, Shen H, Akagi K, Morse HC, Malley JD, et al.
(2002) New genes involved in cancer identified by
retroviral tagging. Nat Genet 32: 166-174. [ FIND THIS ARTICLE ONLINE ]
|