Bin Huang, Jing Guo and Richard Charnigo
DOI: 10.4172/2155-6180.1000e129
Shahab Jolani and Stef van Buuren
DOI: 10.4172/2155-6180.1000194
Estimation in binary longitudinal data by using generalized estimating equation (GEE) becomes complicated in the presence of missing data because standard GEEs are only valid under the restrictive missing completely at random assumption. Weighted GEE has therefore been proposed to allow the validity of GEE's under the weaker missing at random assumption. Multiple imputation offers an attractive alternative, by which the incomplete data are pre-processed, and afterwards the standard GEE can be applied to the imputed data. Nevertheless, the imputation methodology requires correct specification of the imputation model. Dual imputation method provides a new way to increase the robustness of imputations with respect to model misspecification. The method involves integrating the so-called doubly robust ideas into the imputation model. Focusing on incomplete binary longitudinal data, we combine DIM and GEE (DIM-GEE) and study the relative performance of the new method in a case study of obesity among children, as well as a simulation study.
Ming Wang, Junxiang Luo, Haoda Fu and Yongming Qu
DOI: 10.4172/2155-6180.1000195
In diabetes clinical trials, hypoglycemia can be captured. Negative binomial regression is emerging as a standard method for analyzing hypoglycemic events by considering overdispersion. However, in negative binomial regression for hypoglycemic events, variability of the subjects lost to follow up due to dropout is adjusted through an offset parameter, which assumes that dropout is missing completely at random and constant hypoglycemia rate over time. This assumption is vulnerable because dropout may be due to the excessive observed hypoglycemic events and the hypoglycemic event rate may change over time. In addition, the traditional way of using negative binomial regression to analyze hypoglycemic events only compares the counts of hypoglycemic events during a specified period. However, researchers may be interested in comparing hypoglycemic event rates between treatment groups at different time periods to understand the trend over time. Fitting a negative binomial model for each time period ignoring data from other periods may decrease testing power and introduce bias if the assumption of missing completely at random does not hold. We propose piecewise negative binomial regression to incorporate multiple time periods in one model through a generalized linear mixed-effect model. Due to clinical interest, we considered multiple weighting methods to estimate the overall relative rate of hypoglycemia over multiple periods between treatments. Simulations showed that piecewise negative binomial regression performed better than the traditional negative binomial regression in preserving Type I error. As an illustration, piecewise negative binomial regression was implemented in analyzing real data from a Type 2 diabetes clinical trial.
Richard D Riley, Yemisi Takwoingi, Thomas Trikalinos, Apratim Guha, Atanu Biswas, Joie Ensor, R Katie Morris and Jonathan J Deeks
DOI: 10.4172/2155-6180.1000196
Background: When meta-analysing studies examining the diagnostic/predictive accuracy of classifications based on a continuous test, each study may provide results for one or more thresholds, which can vary across studies. Researchers typically meta-analyse each threshold independently. We consider a multivariate meta-analysis to synthesise results for all thresholds simultaneously and account for their correlation.
Methods: We assume that the logit sensitivity and logit specificity estimates follow a multivariate-normal distribution within studies. We model the true logit sensitivity (logit specificity) as monotonically decreasing (increasing) functions of the continuous threshold. This produces a summary ROC curve, a summary estimate of sensitivity and specificity for each threshold, and reveals the heterogeneity in test accuracy across studies. Application is made to 13 studies of protein:creatinine ratio (PCR) for detecting significant proteinuria in pregnancy that each report up to nine thresholds, with 23 distinct thresholds across studies.
Results: In the example there were large within-study and between-study correlations, which were accounted for by the method. A cubic relationship on the logit scale was a better fit for the summary ROC curve than a linear or quadratic one. Between-study heterogeneity was substantial. Based on the summary ROC curve, a PCR value of 0.30 to 0.35 corresponded to maximal pair of summary sensitivity and specificity. Limitations of the proposed model include the need to posit parametric functions for the relationship of sensitivity and specificity with the threshold, to ensure correct ordering of summary threshold results, and the multivariate-normal approximation to the within-study sampling distribution. Conclusion: The joint analysis of test performance data reported over multiple thresholds is feasible. The proposed approach handles different sets of available thresholds per study, and produces a summary ROC curve and summary results for each threshold to inform decision-making.
Derek Shyr and Chung-I Li
DOI: 10.4172/2155-6180.1000198
Power and sample size calculation is an essential component of experimental design in biomedical research. For RNA-sequencing experiments, sample size calculations have been proposed based on mathematical models such as Poisson and negative binomial; however, RNA-seq data has exhibited variations, i.e. over-dispersion, that has caused past calculation methods to be under- or over-power. Because of this issue and the field’s lack of a simulation-based sample size calculation method for assessing differential expression analysis of RNA-seq data, we developed this method and applied it to three cancer sites from the Tumor Cancer Genome Atlas. Our results showed that each cancer site had its own unique dispersion distribution, which influenced the power and sample size calculation.
Tabatabai MA, Kengwoung-Keumo JJ, Eby WM, Bae S, Manne U, Fouad M and Singh KP
DOI: 10.4172/2155-6180.1000199
Background: When outliers are present, the least squares method of nonlinear regression performs poorly. The main purpose of this paper is to provide a robust alternative technique to the Ordinary Least Squares nonlinear regression method. This new robust nonlinear regression method can provide accurate parameter estimates when outliers and/or influential observations are present. Method: Real and simulated data for drug concentration and tumor size-metastasis are used to assess the performance of this new estimator. Monte Carlo simulations are performed to evaluate the robustness of our new method in comparison with the Ordinary Least Squares method. Results: In simulated data with outliers, this new estimator of regression parameters seems to outperform the Ordinary Least Squares with respect to bias, mean squared errors, and mean estimated parameters. Two algorithms have been proposed. Additionally and for the sake of computational ease and illustration, a Mathematica program has been provided in the Appendix. Conclusion: The accuracy of our robust technique is superior to that of the Ordinary Least Squares. The robustness and simplicity of computations make this new technique more appropriate and useful tool for the analysis of nonlinear regressions.
Jingang Miao, Samiran Sinha, Suojin Wang, W Ryan Diver and Susan M Gapstur
DOI: 10.4172/2155-6180.1000197
In modern cancer epidemiology, diseases are classified based on pathologic and molecular traits, and different combinations of these traits give rise to many disease subtypes. The effect of predictor variables can be measured by fitting a polytomous logistic model to such data. The differences (heterogeneity) among the relative risk parameters associated with subtypes are of great interest to better understand disease etiology. Due to the heterogeneity of the relative risk parameters, when a risk factor is changed, the prevalence of one subtype may change more than that of another subtype does. Estimation of the heterogeneity parameters is difficult when disease trait information is only partially observed and the number of disease subtypes is large. We consider a robust semiparametric approach based on the pseudo-conditional likelihood for estimating these heterogeneity parameters. Through simulation studies, we compare the robustness and efficiency of our approach with that of the maximum likelihood approach. The method is then applied to analyze the associations of weight gain with risk of breast cancer subtypes using data from the American Cancer Society Cancer Prevention Study II Nutrition Cohort.
Tabatabai MA, Kengwoung-Keumo JJ, Eby WM, Bae S, Manne U, Fouad M and Singh KP
DOI: 10.4172/2155-6180.1000199
Background: When outliers are present, the least squares method of nonlinear regression performs poorly. The main purpose of this paper is to provide a robust alternative technique to the Ordinary Least Squares nonlinear regression method. This new robust nonlinear regression method can provide accurate parameter estimates when outliers and/or influential observations are present.
Method: Real and simulated data for drug concentration and tumor size-metastasis are used to assess the performance of this new estimator. Monte Carlo simulations are performed to evaluate the robustness of our new method in comparison with the Ordinary Least Squares method.
Results: In simulated data with outliers, this new estimator of regression parameters seems to outperform the Ordinary Least Squares with respect to bias, mean squared errors, and mean estimated parameters. Two algorithms have been proposed. Additionally and for the sake of computational ease and illustration, a Mathematica program has been provided in the Appendix.
Conclusion: The accuracy of our robust technique is superior to that of the Ordinary Least Squares. The robustness and simplicity of computations make this new technique more appropriate and useful tool for the analysis of nonlinear regressions.
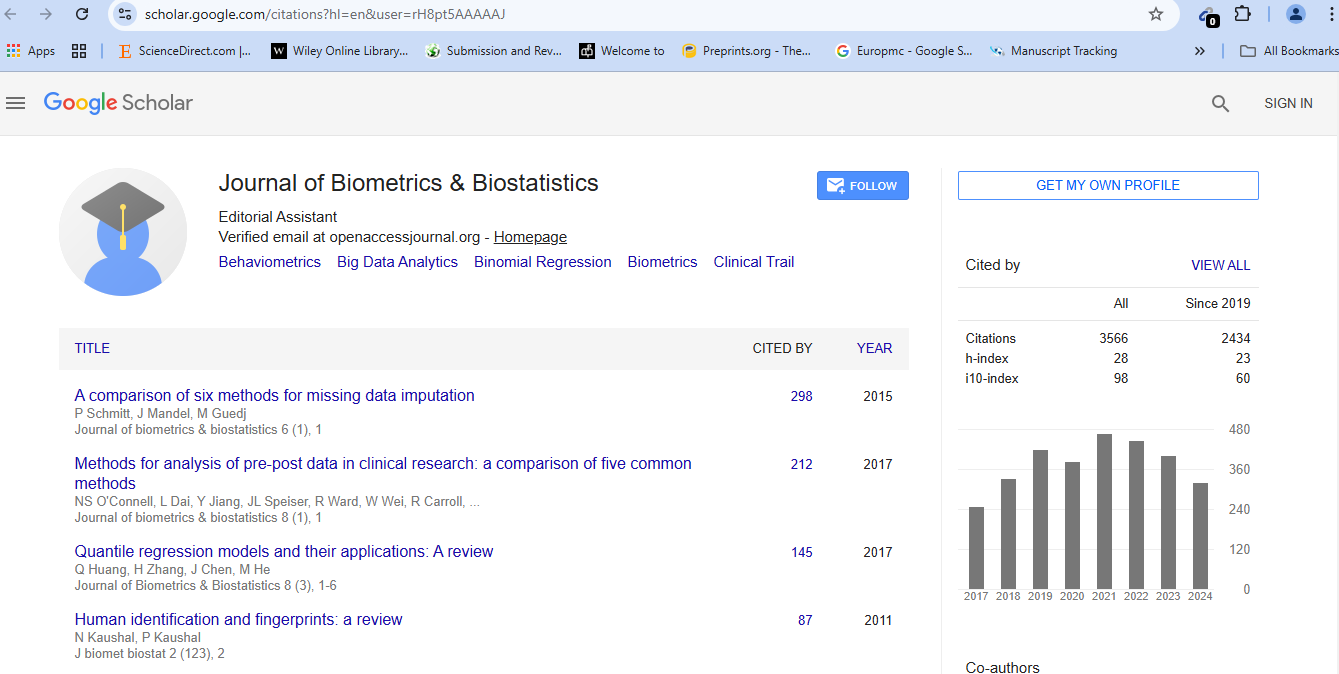
Journal of Biometrics & Biostatistics received 3254 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 