|
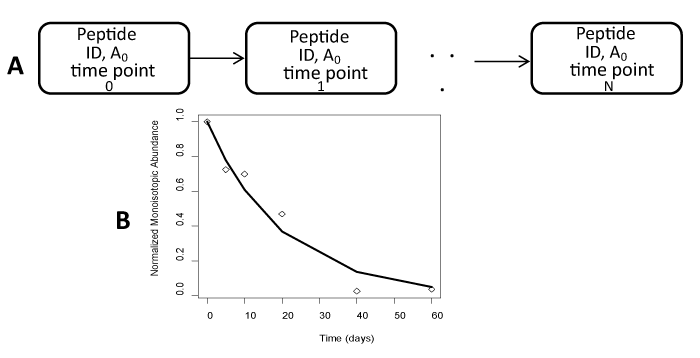
| Figure 1: Time-course progression of data processing and isotopomer computation in proteome dynamics experiments with heavy water labeling. In the first step, A., for every time point, a peptide is identified from using tandem mass spectra, precursor mass and a protein sequence database. To avoid statistical problems associated with missing data, only peptides that are consistently identified in all time-course experiments are used. Relative abundance values, A0, of the monoisotopic proportions are determined from Brauman’s least squares distribution. In the second step, B., the monoisotopic proportions, A0, at every time are fit to an exponential time-dependent decay function to determine peptide turnover rate. |