|
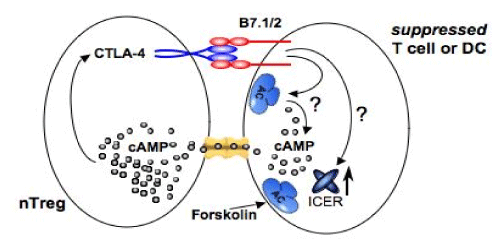
| Figure 6: Cyclic AMP underpins suppression by nTreg cells. Upon TCR activation (not shown), CTLA-4 is deployed to the surface of nTreg cells, and a high-affinity CTLA-4/B7 interaction in synergy with intercellular gap junction formation (in yellow) leads to cAMP formation [51,52,53,74] and subsequent cAMP influx followed by the immediate, early induction of ICER in TCR-activated Foxp3neg Tcons [44]. Analogous effect could be achieved by direct activation of Adenylyl Cyclase (AC) by forskolin or inhibition of Phosphodiesterases (PDEs) responsible for degradation of cAMP e.g. by Rolipram [55]. In response to cAMP-ICER is induced (after 2-4 h of delay, necessary for ICER synthesis) in the Foxp3neg Tcons [44] and later ICER protein is enforced to the nucleus in response to cAMP where it attenuates IL-2 expression, induced by TCR activation [55]. TCR-activated Tcons promote CTLA-4 and B7 expression in cAMP-dependent fashion [74,75] during delay in ICER expression (ICER is absent and/or cytosolic) [55]. This could lead to ‘processive’ ICERmediated transcriptional attenuation of IL-2 expression by CTLA-4/B7 interaction in ‘infectious’ manner in the next neighboring activated Foxp3neg Tcons. When ICER is in the nucleus (whether this is a result of direct, intracellular Foxp3 expression in nTregs, and/or cAMP influx in suppressed Tcons, or both), autonomous CTLA-4 signaling inhibits ERK and thus protects ICER from ERK-mediated phosphorylation, subsequent ubiquitination, and nuclear de-localization [55]. In this model, nTreg cells modulate activity of autoreactive Tcons and/or Dendritic Cells (DCs) through high-affinity CTLA-4/B7 and Class II-TCR interactions (not shown). |