|
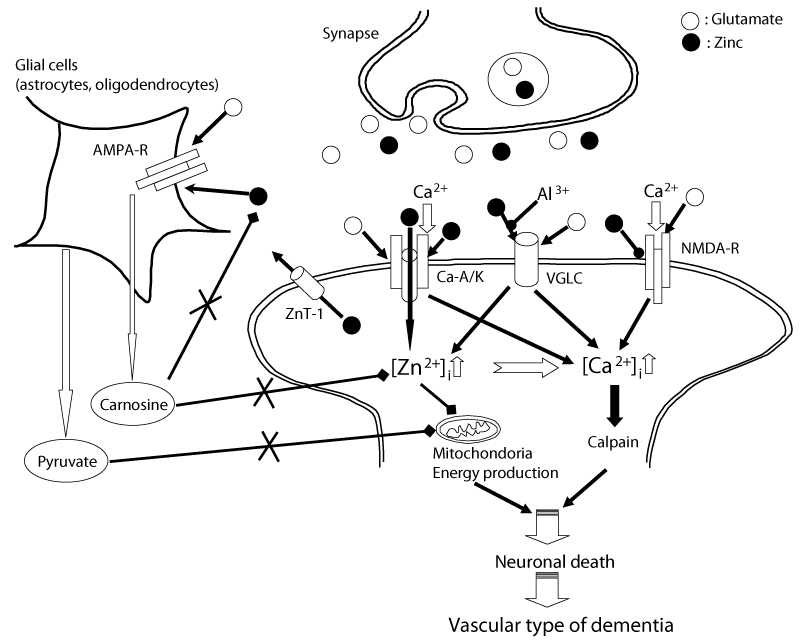
| Figure 6: Hypothetical scheme of Zn neurotoxicity. Zn (Zn: closed circle) coexists with glutamate (Glu: open circle) in presynaptic vesicles and is secreted upon neuronal excitation. Under normal conditions, secreted Zn binds to NMDA-type glutamate receptors (NMDA-R) and modulates postsynaptic excitability. Carnosine is synthesized in glial cells and is secreted in response to stimulation by glutamate and Zn; it protects neurons from glutamate-Zn neurotoxicity. This feedback pathway contributes to Zn homeostasis. Concentrations of intracellular Zn ([Zn2+]i) are also maintained by ZnT-1, a zinc transporter. Pyruvate is also released from glial cells and protects neurons from mitochondrial energy deficits caused by Zn. However, under pathological conditions such as transient global ischemia, large amounts of both glutamate and Zn are released into synaptic clefts. Zn potentiates the expression of Ca2+-permeable AMPA/kainate-type glutamate receptor (Ca-A/K) channels. Zn is translocated into postsynaptic target neurons through Ca-A/K channels or other pathways such as voltage-gated L-type Ca2+ channels (VGLC) or NMDA-R. Increased [Zn2+]ii inhibits numerous enzymes, including mitochondrial respiratory enzymes, and thus causes energy depletion. Meanwhile, excess glutamate also increases [Ca2+]i. The increase of Ca-A/K channels and the increased [Zn2+]i both contribute to the increase in [Ca2+]i. Increased [Ca2+]i consequently triggers various apoptotic pathways, including the activation of calpain. Carnosine levels are decreased in aged bodies. Therefore, the protective effects of carnosine in the hippocampus or cerebral cortex might be insufficient in cases of pathological conditions under the aged brain. Dyshomeostasis of Zn and Ca eventually causes delayed neuronal death after transient global ischemia; it ultimately leads to the pathogenesis of VD. |