|
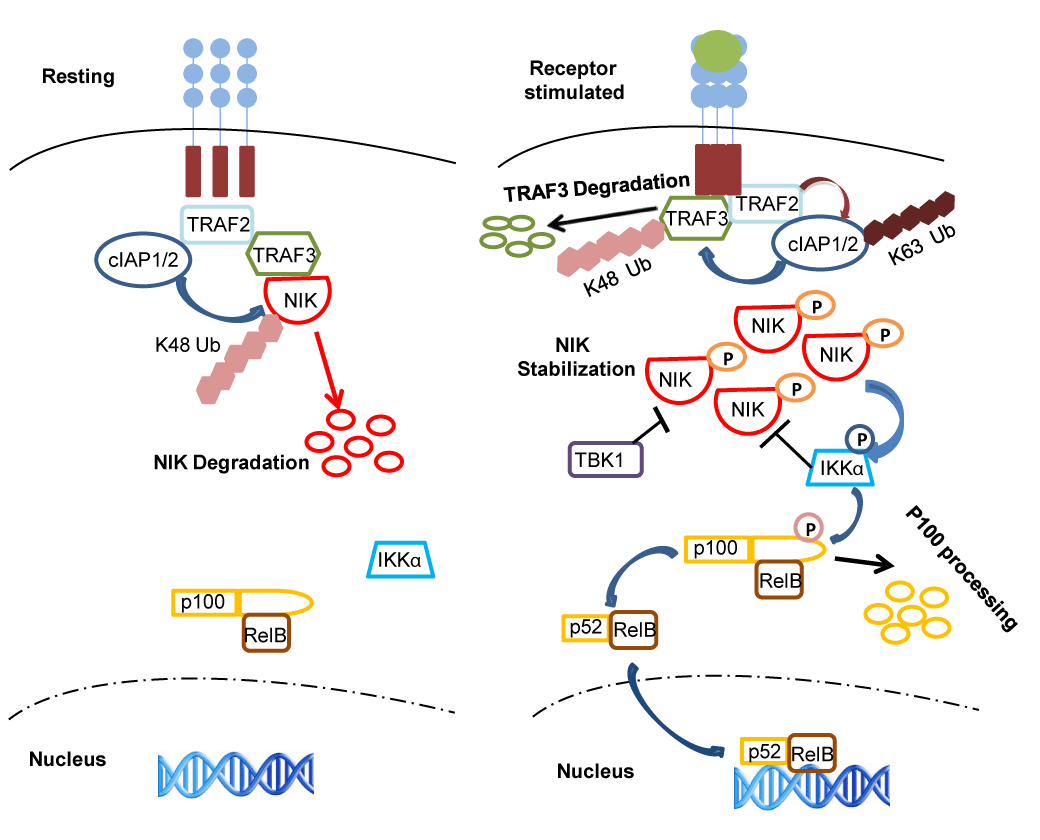
| Figure 1: Regulation of the alternative NF-κB pathway in resting vs receptor stimulated cells. A tight control on NIK stability is essential to achieve controlled activation of the alternative NF-κB pathway upon activation of different receptors. In resting cells, a complex comprising cIAP-TRAF2-TRAF3-NIK induces constant degradation of NIK making its levels invisible. In this complex TRAF3 functions as a bridging factor between the cIAP-TRAF2 complex and NIK enabling cIAP to mediate K-48 linked ubiquitination of NIK leading to its degradation in a proteasome-dependent manner. In receptor-stimulated cells however, NIK escapes the ubiquitination by the cIAPTRAF2- TRAF3 complex due to rapid degradation of TRAF3 by the proteasome machinery. Upon receptor stimulation, TRAF2-dependent K63-linked ubiquitination of cIAP seems to mediate cIAP-dependent K-48 linked ubiquitination of TRAF3 leading to the proteasome-dependent degradation of the latter. Once the TRAF3 levels drop below a threshold, NIK can no longer interact with the TRAF2-cIAP complex. As a consequence NIK gets stabilized and gets activated presumably due to transautophosphorylation. Activated NIK phosphorylates IKKa, which in turn phosphorylates p100 resulting in p100 processing and nuclear localization of RelB-p52 and their DNA binding. In receptor stimulated cells, IKKa and the kinase TBK1, phosphorylate NIK and induce its degradation to dampen the alternative NF-κB activity. |