Ibtessam Ramzi Hussein, Bassiouni R, Chaudhary A, Al Malki and Al Qahtani M
We report on a new case with developmental delay, dysmorphic features, holoprosencephaly that showed deletion in long arm of chromosome 21 (21q22.13-q22.3). The patient is a male 3 months old presented with frontonasal dysplasia, scoliosis, abnormal ears, VSD, hypospadias, undescended testis. MRI has shown holoprosencephaly and agenesis of corpus callosum. Array-comparative genomic hybridization using the Agilent 2×400 oligoarray showed an interstitial deletion in chr21q22.13-q22.3, (start-end: 36,854,967-46,006,008 bp) deletion size is 9 Mb (9,151,042 bp) and includes 75 genes (Data base of genomic variants, hg18). The deletion was found to be maternal in origin. The findings from this report underscore the role of the genes at chromosome 21q22.13-q22.3 in brain development and indicate the usefulness of array-CGH in identification of the deletion size and detection of genes that could be correlated to the patient’s phenotype.
PDFShare this article
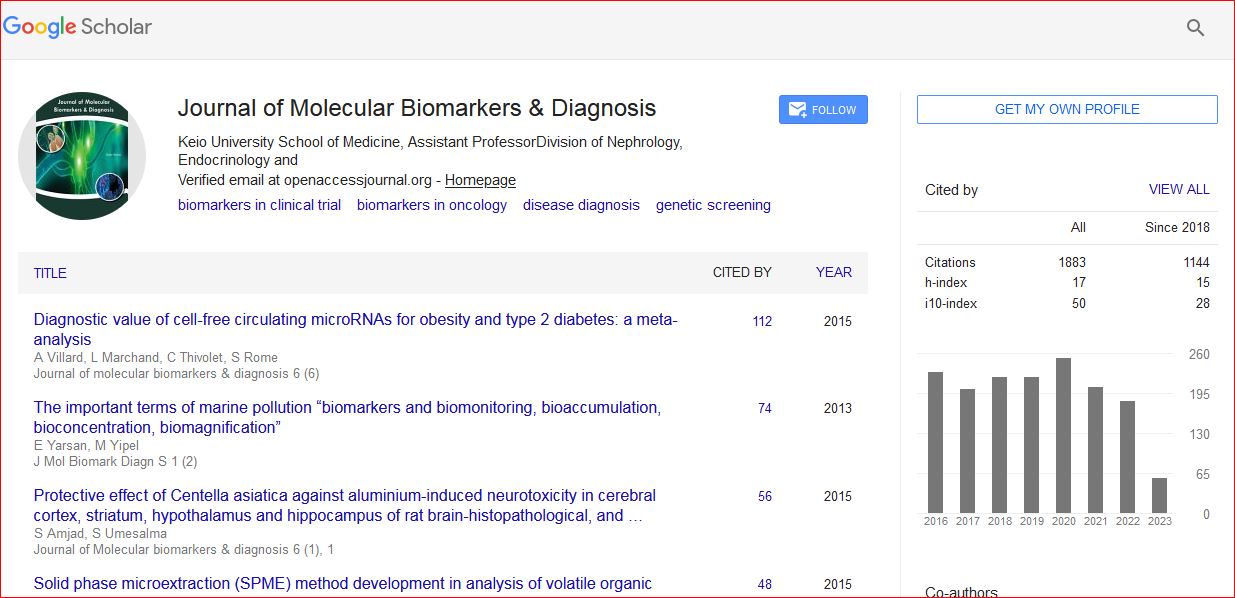
Journal of Molecular Biomarkers & Diagnosis received 2054 citations as per Google Scholar report