Leila Chaouch, Imen Darragi, Emna Barkaoui, Slim Ben Ammar, Dorra Chaouachi, Imen Boudrigua and Salem Abbes
Introduction: Gilbert syndrome (GS) is due to a defect in uridine diphosphate glucuronosyl transferase (UGT1A1) gene and belongs to the group of the most common human metabolic disorders and is characterized by an elevated level of bilirubin in blood serum. Genotyping functional polymorphisms in UGT1A1 gene is an important step in the determination of the etiology of free hyperbilirubinemia of unknown origin. Herein, we aimed to explain the genetic profile and the clinical variability of this disease in Tunisian patients.
Material and methods: We explored a total of 30 subjects including 7 unrelated isolated cases and 23 subjects distributed to 7 families. The exploration of these subjects included the genotyping of two Functional polymorphisms of UGT1A1 gene associated with GS namely: A(TA)nTAA and G71R, the bilirubin level, the hematological parameters, the determination of glucose-6-phosphate dehydrogenase activity (G6PD) and echo dense images within the gall bladder with acoustic shadowing or gravitational change in position.
Results: The exploration of the hematological parameters showed that all subjects enrolled in this study are out of hemolytic disease with the exception of two individuals, it is a father and his son who have a profile of a β thalassemia minor. The exploration of G6PD activity showed that all patients presented normal values. All patients presented hyperbilirubinemia among whom some patients presented benign asymptomatic jaundice, two patients presented kernicterus and 4 patients presented cholelithiasis. The genetic profile studied of UGT1A1 revealed that the mutant allele A of the G71R polymorphism was absent for all subjects and that the genotype (TA)7/(TA)7 was associated with hyperbilirubinemia and with GS.
PDFShare this article
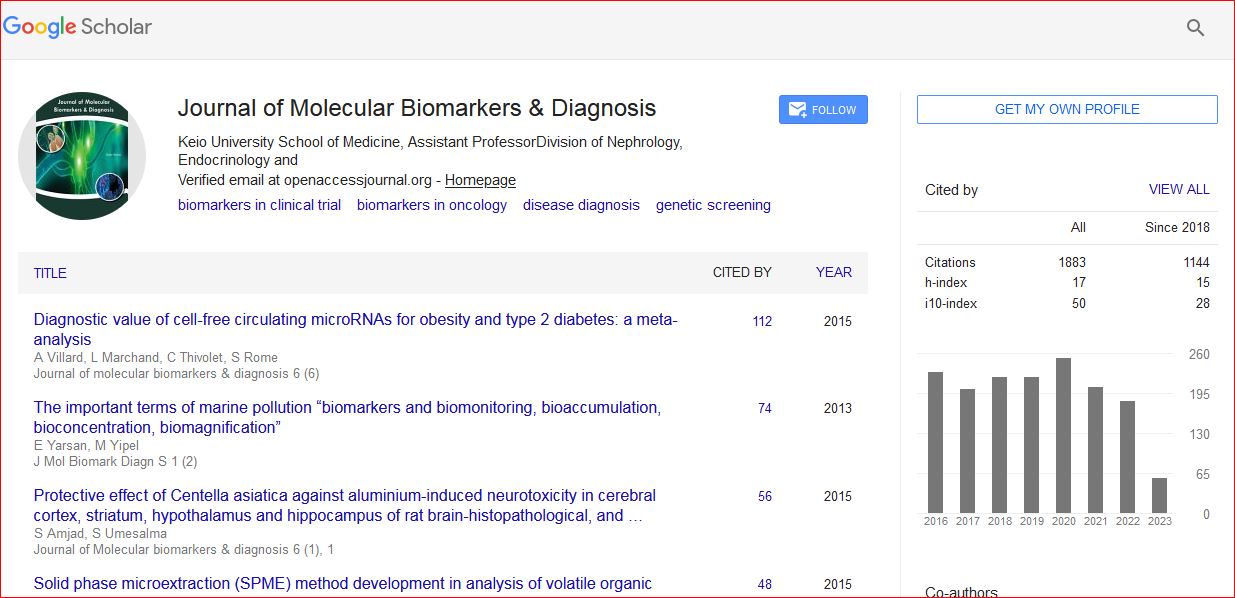
Journal of Molecular Biomarkers & Diagnosis received 2054 citations as per Google Scholar report