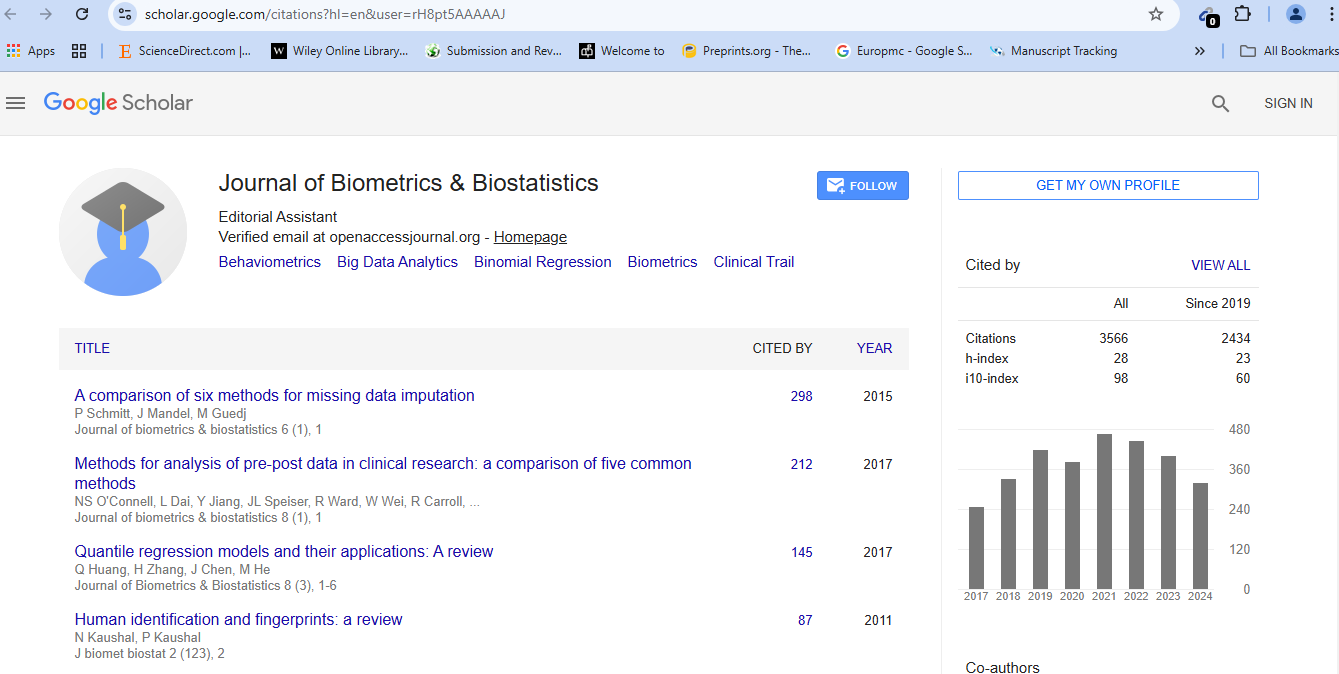
Contaminated Chi-Square Modeling and Large-Scale ANOVA Testing
Abstract
Richard Charnigo, Feng Zhou and Hongying Dai
We propose a convenient moment-based procedure for testing the omnibus null hypothesis of no contamination of a central chi-square distribution by a non-central chi-square distribution. In sharp contrast with likelihood ratio tests for mixture models, there is no need for re-sampling or random field theory to obtain critical values. Rather, critical values are available from an asymptotic normal distribution, and there is excellent agreement between nominal and actual significance levels. This procedure may be used to model numerous chi-square statistics, obtained via monotonic transformations of F statistics, from large-scale ANOVA testing, such as that encountered in microarray data analysis. In that context, modeling chi-square statistics instead of p-values may improve detection of differential gene expression, as we demonstrate through simulation studies, while also reducing false declarations of the same, as we illustrate in a case study on aging and cognition. Our procedure may also be incorporated into a gene filtration process, which may reduce type II errors on genewise null hypotheses by justifying lighter controls for Type I errors.
PDF