David Fardo
Scientific Tracks Abstracts: J Biomet Biostat
The rapid advances of cost-efficient genetic sequencing technologies have made the study of genetic associations with complex diseases commonplace. Broadly speaking from a study design perspective, these studies can be distinguished by whether or not families (i.e., related individuals) are recruited. It is not uncommon for a particular disease to have substudies designed that collect either all unrelated individuals or all families. When this is the case, the choice of how to combine the evidence for association across studies can greatly influence statistical power. Various study design aggregation approaches have been developed, most commonly in the context of common variants. Recently developed Next Generation Sequencing (NGS) technologies have made it feasible to assess association between multiple rare variants and disease. This, in combination with motivation to target the so-called �missing heritability� from Genome-Wide Association Studies (GWAS), has made the study of rare variation particularly attractive. We present here a unified method to aggregate population and family data using GWAS or NGS data and assess its performance using simulation.
David Fardo received his Ph.D. in Biostatistics from Harvard University in 2008. He is currently an Assistant Professor of Biostatistics at the University of Kentucky where has developed a statistical genetics curriculum. His statistical interests focus on developing and applying methodologies for understanding how genetic variation relates to complex human diseases.
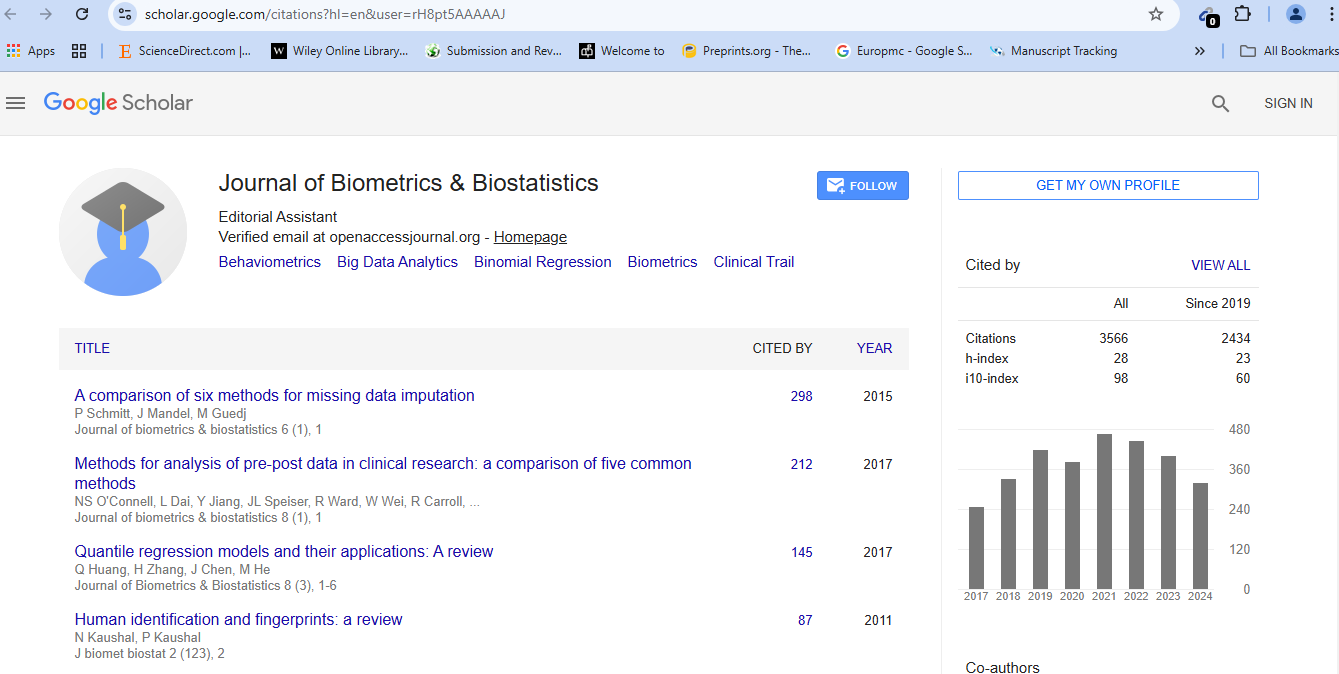
Journal of Biometrics & Biostatistics received 3254 citations as per Google Scholar report

 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi 