Muraro E, Gianesello L, Priante G, Comacchio A, Carraro G, Naso A, Anglani F, Valente M and Del Prete D
Background: Hyperoxaluria may be either inherited or acquired. Primary Hyperoxaluria (PH) is a rare autosomal recessive disease characterized by increased endogenous oxalate production and accumulation in renal and extrarenal tissues. The excess oxalate is excreted in the urine and frequently patients with PH present signs or symptoms related to kidney stones and progressive nephrocalcinosis. Here we present a case of a young man with an unexplained progressive renal failure, without symptoms of nephrolithiasis or nephrocalcinosis. Renal biopsy examination was performed to clarify renal dysfunction. Kidney biopsy showed a glomerular and tubulo-interstitial nephropathy by oxalate deposits. Genetic testing was used to confirm histopathological evaluation, demonstrating the c.731T>C mutation at exon 7 of AGTX gene. Conclusions: This case of PH type 1 was peculiar for the clinical presentation (renal failure without evidence of urolithiasis or nephrocalcinosis) and for the glomerular histopathological aspect of oxalate deposition. To our knowledge, this is the first demonstration of CaOx deposition in the glomeruli to be reported in the literature. The histopathological diagnosis enable us to study in deep the bio-humoral profile of the patient and to reach an accurate diagnosis.
PDFShare this article
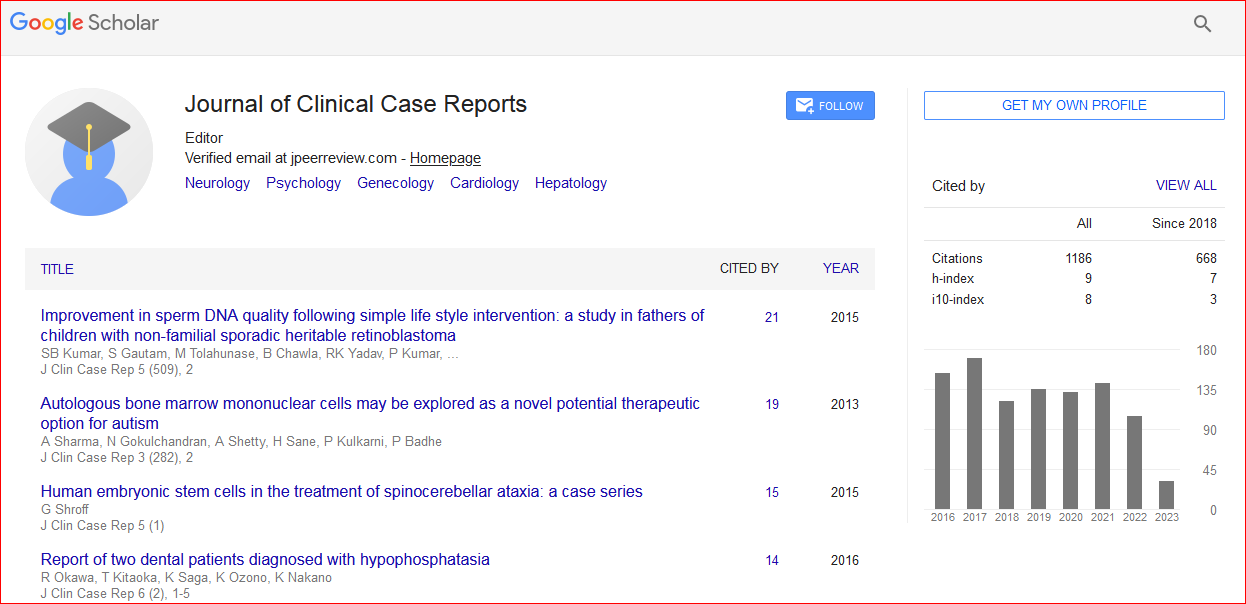
Journal of Clinical Case Reports received 1295 citations as per Google Scholar report