| Research Article |
Open Access |
|
| Yu-Lun Kuo1#, Peter Mu-Hsin Chang2,3#, Yu-Wen Liu4#, Po-Hsu Chen5, Pei-Ying Lee4, Ren-Shyan Liu6, Jin-Mei Lai7, Yin-Jing Tien5, Yu-Chung Wu 8, Li-Jen Su9, Cheng-Yan Kao1,10, Chun-Houh Chen5* and Chi-Ying F. Huang3,4* |
| 1Department of Computer Science and Information Engineering, National Taiwan University, Taipei, Taiwan |
| 2Division of Hematology and Oncology, Department of Medicine, Taipei Veterans General Hospital, Taipei, Taiwan |
| 3Institute of Clinical Medicine, National Yang-Ming University, Taipei, Taiwan |
| 4Institute of Biopharmaceutical Sciences, National Yang-Ming University, Taipei, Taiwan |
| 5Institute of Statistical Science, Academia Sinica, Taipei, Taiwan |
| 6Department of Nuclear Medicine, Taipei Veterans General Hospital, Taipei, Taiwan |
| 7Department of Life Science, College of Science and Engineering, Fu Jen Catholic University, New Taipei City, Taiwan |
| 8Division of Thoracic Surgery, Department of Surgery, Taipei Veterans General Hospital, Taipei, Taiwan |
| 9Institute of Systems Biology and Bioinformatics, National Central University, Jhongli City, Taiwan |
| 10Graduate Institute of Biomedical Electronic and Bioinformatics, National Taiwan University, Taipei, Taiwan |
| #These authors contributed equally to this study |
| *Corresponding authors: |
Chi-Ying Huang
Institute of Clinical Medicine
National Yang-Ming University
No. 155, Sec. 2, Linong St
Taipei 112, Taiwan
Tel: +886- 228267904
Fax: +886-228224045
E-mail: cyhuang5@ym.edu.tw |
| |
Chun-Houh Chen
Institute of Statistical Science
Academia Sinica, 128 Academia Road Sec.2
Nankang Taipei 115, Taiwan
Tel: +886-227835611 ext. 407
Fax: +886-227831523
E-mail: cchen@stat.sinica.edu.tw |
|
| |
| Received December 14, 2011; Published June 20, 2012 |
| |
| Citation: Kuo Y, Chang PM, Liu Y, Chen P, Lee P, et al. (2012) In silico Therapeutic Drug Screening for Reversing the Lung Adenocarcinoma Overexpressed Gene Signatures. 1: 101. doi:10.4172/scientificreports.101 |
| |
| Copyright: © 2012 Kuo Y, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| Lung cancer is the leading cause of mortality in the world. However, the urgent demand of new drugs for the treatment of lung cancer and the rapidly rising costs of drug development support efforts to explore methodology addressing these issues. Here we designed various gene signature selection methods via expression datasets generated from human lung adenocarcinoma tumor and adjacent non-tumor tissues to query the Connectivity Map (CMap), which hosts 6,100 drug-mediated expression profiles. We hypothesized that if a drug signature could reverse, at least in part, the gene expression signature of lung adenocarcinoma, it might have the potential to inhibit dysregulated pathways and thereby treat lung cancer. To test this hypothesis, 62 out of the 95 examined drugs with anti-tumor activities were validated via MTT and/or clonogenic assays using lung cancer cell lines and through a PubMed search. Subsequently, 9 functional categories, such as DNA demethylating agents, anti-psychotic and antiinflammation, were selected to classify 62 in vitro validated drugs. A subset of gene signatures embedded in these 62 drug-mediated expression profiles from the CMap was uncovered, suggesting the possible reversal connection between drug and patient gene signatures. There were 89 differentially expressed probesets that produced a reverse signature, including 12.5% that follow the up-regulated (patient) to down-regulated (CMap) pattern and 16.6% with the reverse down-regulated (patient) to up-regulated (CMap) pattern. Such bioinformatics analysis makes functional connections among disease, genetic perturbation, and drug action, resulting in the systematic identification of potential therapeutic drugs for clinical use. |
| |
| Keywords |
| |
| Non small cell lung cancer; Adenocarcinoma; Pyrvinium; Chlorpromazine; Connectivity Map |
| |
| Introduction |
| |
| Lung cancer is the leading cause of death in the United States [ 1] and Taiwan [2]. According to the World Health Organization (WHO) classification, lung cancer can be divided into two major classes: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC accounts for more than 85% of all lung cancer cases, and adenocarcinoma is the most common subtype. There is a large heterogeneity among lung adenocarcinoma patients and the response to chemotherapy is relative poor in the advanced stage. For example, gefitinib, the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI), is only useful for East Asian patients with a higher prevalence of activating EGFR mutations [3-6]. Other targeted agents for EGFR wild-type patients have not been systemically screened. Therefore, the search for novel targeted drugs is an important and challenging issue. |
| |
| Using microarray profiling, one can identify a handful of differentially expressed genes from NSCLC tumor and adjacent nontumor tissue in each patient. The fundamental challenge is to establish the relationships among these carcinogenic associated genes and potential drug actions. One possible solution is via the “Connectivity Map” (https://www.broad.mit.edu/cmap/) [7,8]. The CMap system contains 6,100 microarray datasets from 4 cancer cell lines (MCF7- breast cancer, PC3-prostate cancer, HL60-leukemia, SKMEL5- melanoma) treated with 1,309 small molecular agents. Lamb et al. [ 7,8] postulated that the disease associated gene signatures could be compared to the CMap drug signature profiles to reveal potential drug lists despite that the profiles are in different cell lines. In fact, potential new treatments for cancers have been successfully identified via the CMap, including acute leukemia, colon cancer, hepatocellular carcinoma, neuroblastoma, NSCLC, and renal cell carcinoma, and it has also led to the elucidation of mechanisms involved in the tumorigenesis and/or pathogenesis of different cancers [9-11]. |
| |
| One criterion for use of the CMap is to include a pairwise array. However, to the best of our knowledge, few tumor/adjacent non-tumor pairwise arrays are available for NSCLC. In addition to our previously generated pairwise samples (referred to as the YM dataset) [12], we used two additional pairwise arrays available online, including the partial dataset from the EAGLE (Environment And Genetics in Lung cancer Etiology, https://eagle.cancer.gov/) study [13] from Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) and the pairwise gene signature is available from ArrayExpress (https://www.ebi.ac.uk/arrayexpress/). |
| |
| Here, we generated an additional 50 pairwise arrays for NSCLC and employed the differential gene expression profiles from the tumor compared with the adjacent non-tumor lung tissues in each patient to create a gene-model to query the CMap database to identify potential novel targeted drugs, followed by the systematic evaluation of the in vitro anti-tumor effects of the potential drugs. To identify the reverse signature probesets, we used 705 instances (chips) from 62 drugs of 9 functional categories to determine the differentially expressed probesets that have a reverse signature from up-regulated (patient) to down-regulated (CMap) and down-regulated (patient) to up-regulated (CMap) patterns, respectively. For example, p21, which has been observed significantly down-regulated in NSCLC tumors, was up-regulated after trichostatin A treatment and resulted in tumor shrinkage in the NOD/SCID mice. This comprehensive analysis suggests that we could use individualized gene signatures to identify novel target agents, which may provide therapeutic regimens toward personalized medicine in the future. |
| |
| Materials and Methods |
| |
| Clinical samples for microarray analysis |
| |
| We analyzed a total of 172 samples from three cohorts of patients with NSCLC via microarray analysis (Supplemental Table S1). Dataset 1 included two lung adenocarcinoma microarray datasets used in this study. First, from our previous study, a total of 38 pairwise samples from 19 patients (GSE7670) were used for analysis via the Affymetrix HG-U133A chip [12]. Second, an additional 50 pairwise surgical samples from 25 patients (GSE27262, currently private) were subjected to analysis via the HG-U133 plus 2.0 chip. Among these data are 66 stage-IA/IB and 22 stage-III/IV pairwise samples. The study protocol was approved by the Ethics Committee at Taipei Veterans General Hospital. All patients gave informed consents and signed the consent form individually (VGH IRB No.: 95-06-21A). Dataset 2 (GSE10072) was obtained from Landi et al. [13] and included 30 stage-IA/IB (15 patients) and 18 stage-III/IV (9 patients) pairwise samples using the HG-U133A chip. Dataset 3 (ArrayExpress ID E-TABM-15) published by Shula Blum and included 36 unknown stage (18 patients) pairwise samples using the Affymetrix HG-U133A chip. The details of the patient characteristics are summarized in Supplemental Table S2-S4 and Supplemental method. |
| |
| In silico drug screening |
| |
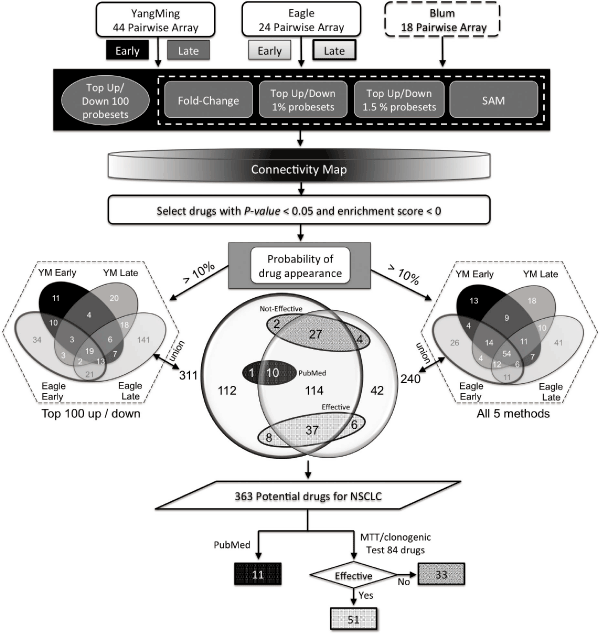
| The “up” or “down” differential probesets of individualized gene signatures from tumor/adjacent non-tumor in each patient was used to query the CMap. The potential drug list for each patient was selected based on the frequency of appearance among patients. Drugs that appeared with top-ranked frequency when querying the CMap with significantly negative scores were listed and compared with each other. The flowchart of analyzing the 172 microarray data points and the in silico drug screening is shown in Figure 1. |
| |
|
|
Figure 1: Flowchart of array analysis and appearance of potential drugs in NSCLC. The YangMing and EAGLE microarray datasets, consisting of early and late stage lung adenocarcinoma patient information, were used for data analysis, whereas array datasets from the Blum study did not have detailed information and were used for validation. Five different methods (top up/ down 100 probesets, fold-change, SAM, top 1% and 1.5% ranked up/down probesets) were used respectively to create differential gene lists of tumor/ adjacent non-tumor tissue for querying the Connectivity Map. Only drugs with a p value of less than 0.05 and a negative enrichment score were retained. Two drug lists were generated. First, we used the probability of drug appearance (> 10%) among each studied population to select drugs and a total of 311 drugs were selected using the top up/down 100 probesets as query. Second, using the probesets generated from all five methods to perform the same analysis resulted in the identification of 240 drugs. A Venn diagram represents the number of drugs found between the top up/down 100 probesets and all five methods. There are 188 drugs that overlap between the two approaches, including 10 drugs that have been previously studied. In this study, 84 drugs were selected based on the availability of well known scientific names and manufacturers from all 363 union drugs and subjected to validation via MTT/ clonogenic assay. There are 62 effective drugs, which include 11 drugs from the literature, of the 95 tested potential drugs for NSCLC. |
|
| |
| Gene Selection |
| |
| Because the best method to query the CMap is unclear, we designed five methods to select differentially expressed genes as inputs. Moreover, the “up” and “down” probesets used for querying the CMap should not be over a total of 1000 due to the limitation of the CMap computational system. Therefore, all five methods were limited to less than 1000 probesets in each query. The first method selected 100 probesets with the highest absolute ratios from our early- (stage IA/IB lung ADC) and late- (stage III/IV lung ADC) stage paired microarrays. Therefore, signatures including 100 up- and 100 down-regulated probesets were used to query the CMap. The second method and third method included the top 1% or 1.5% ranked up- and down-regulated probesets on Affymetrix HG-U133A microarray. These methods can identify a total of 444 or 668 probesets, respectively, as the differential expressed signatures. Next, fold-change, which depends on the expression value of gene relative to the control expression profile, was used to identify significant expressed probesets. Finally, Significance Analysis of Microarrays (SAM), a statistical algorithm that can identify significant genes by specific t-test [14] (https://www-stat.stanford. edu/~tibs/SAM), was used. |
| |
| Statistical procedure and gene expression profiling for 9 functional categories of drugs |
| |
| To investigate the relationships among the drugs, their regulated genes and lung cancer patient signatures, we classified these 62 effective drugs into 9 functional categories, including antibiotics, anti- inflammation, anti-parasites, Ca2+ modulators, cardiovascular drugs, chemotherapeutic drugs, DNA demethylating agents, phenothiazine anti-psychotics, and others. There are (6, 10, 6, 3, 8, 11, 3, 5, 10) drugs and (23, 200, 29, 11, 33, 95, 197, 76, 41) instances for each functional category, respectively, in the CMap database. Thus, the original CMap expression profile (U133A chip) contains 22,283 gene expressions (ratio of probeset) each for 705 instances from 62 drugs within 9 functional categories. Several statistical procedures (i.e., the size of standard deviation, analysis of variance (ANOVA), and the pairwise t-test) combined with fold-changes were employed to select gene profiles representing the 9 drug functions, but most were dominated by functional categories with larger number of drugs and instances, such as DNA demethylating agents and anti-inflammation. For each drug, 22,283 drug medians (one for each probeset) of expression ratios were calculated across all instances within each drug. Then, for each functional category, 22,283 functional medians of expression ratios were calculated across all drug medians within that function. One hundred probesets with the largest (or smallest) functional median ratios that were larger than log2 (1.5) = 0.585 (or smaller than -log2 (1.5) = -0.585) were selected as the up-regulated (or down-regulated) genes for that functional category. All of the probesets that corresponded to multiple gene symbols or no gene symbol were excluded from the final analysis. 624 probesets survived this screening procedure with Table 1 summarizing these 265 up-regulated and 359 down-regulated probesets elected for the 9 functional categories. Two datasets were then prepared for later analyses. The drug-level dataset has median of expression ratios for each of the 624 (probeset) by 62 (drug) combinations and the function-level dataset has median (of medians) of expression ratios for each of the 624 (probeset) by 9 (function) combinations. |
| |
|
|
Table 1: Number of representative probesets selected for the nine functional categories. |
|
| |
| Cell culture, MTT™ cell viability test, clonogenic assay, and animal model |
| |
| All cell-culture-related reagents and procedures are in supplemental method. Human lung cancer cell lines A549 and H460 were purchased from the American Type Culture Collection/ Bioresource Collection and Research Center (BCRC) (Taiwan). These cells have performed STR-PCR profile at BCRC. A14 was a derivative of A549 cells stably selected with a p53 shRNA construct [15]. Human lung adenocarcinoma cell lines, CL1-0 and CL1-5 [16], were kind gifts from Dr. Pan-Chyr Yang. H1299 stable clones (transfected with EGFRWT (wild-type) and EGFR-L858R mutant) were kindly provided by Chen et al. [17]. Cell viability was determined using an MTT assay and clonogenic assay were described in supplemental method. In vivo microPET imaging of overexpression of p21 induced by trichostatin A in p21-HSV1-tk expressed H1299 animal tumor model [18] was also described in Supplemental method. |
| |
| Results |
| |
| Identification of 363 potential drugs for NSCLC via the CMap |
| |
| The use of global gene expression profiling of surgically excised tumor samples to identify prognostic signatures or molecular classification of lung tumors often generates lists of hundreds of genes and it is difficult to uncover novel drug targets [19-21]. Therefore, using a batch of genes to query the CMap may not only allow multiple targets to be considered simultaneously but also may identify potential new drugs. For our first analysis, we selected the top 100 up- and down-regulated probesets with the highest absolute ratio to serve as differential gene signatures of individual patients from all early and late stage pairwise microarrays of the first two datasets. We then used the CMap to identify potential drugs with p value less than 0.05 and a negative enrichment score. With the probability of drug appearance (> 10%) among each studied population as selection criteria, a total of 311 drugs were selected by the top 100 up/down probesets. |
| |
| However, there is no standard approach to query the CMap due to different input gene list, which may result in distinct analysis results. Unlike the traditional gene signatures selected with the most differentially common sets for predicting the cancer prognosis (e.g., two-sample t-test), we employed 4 additional methods (fold-change, top 1% ranked, top 1.5% ranked, and SAM) to generate different input gene lists to broadly cover potential drugs for lung cancer. Among them, SAM is the well-known statistics method for the selection of significant genes. Fold change is a method for selection of genes based on the degree of expression ratio (e.g., 2-fold), which may prevent the selection of genes without significant expressive change. Additionally, considering the limitation of query gene numbers in the CMap platform, we used the top 1% and 1.5% of up and downregulated genes to present the differential signatures. Next, we selected the probesets from each of 5 methods to perform the same approach, respectively. As a result of data fusion, 240 drugs with an appearance of at least 10% were selected. From the analysis of this study, the top 100 up/down showed the highest intersection ratio (top 100 up/down, top 1% ranked, top 1.5% ranked, fold-change, and SAM with 60.5% vs. 56.1% vs. 56.4% vs. 46.5% vs. 42.4%, respectively) with fusion to the total of the five populations (Figure 1 and Supplemental Table S5). We are not able to use all five or more methods for the generation of the differentially expressed signature for every experiment due to the time constraints of gene selection. Consequently, we choose the top 100 up/down method as our standard selection criteria for current and future work with the CMap. As shown in the middle of Figure 1, the Venn diagram represents the intersection of 311 drugs from the top 100 up/down probesets and 240 drugs generated from fusion of all five methods. There are 188 drugs that overlap between the two analyses and 363 potential drugs as a union of these two analyses. |
| |
| |
| Validation of 62 potential drugs via PubMed search and biochemical assays |
| |
| Of these 363 potential drugs, several drugs have been well studied in regards to their in vitro anti-cancer cell effects, such as LY-294002, resveratrol, and alvespimycin. These “familiar drugs” were subjected to search of the PubMed medical literature database using the search term of each drug. We then screened the resulting abstracts for relevance or involvement in “cancer” and/or “lung cancer”. As a result, 11 drugs have more publications related to cancer and lung cancer. These were considered as effective drugs and were excluded from further experimental analysis. Interestingly, 10 of the 11 drugs belonged to the 188 drugs that overlapped between the two analyses (Figure 1). |
| |
| To further validate our analysis, we selected 84 potential drugs ( Table 2) that based on the availability of well-known scientific names and manufacturers. We tested the cytotoxicity of 84 drugs from the CMap in five lung cancer cell lines via an MTT and/or a clonogenic assay. To determine the role of p53 in the cytotoxic effect, we examined the drug effects in the A549 and the A549-p53 shRNA stable clone cell lines [15]. To examine the role of EGFR in drug cytotoxicity, we used EGFR over-expressing H1299 cells, including wild-type and mutated EGFR [17]. |
| |
|
|
Table 2: Potential drugs identified from 3 lung adenocarcinoma datasets including stage I and stage III/IV. There are nine functional groups for the 62 predicted lung cancer drugs, which have been validated in vitro by MTT/clonogenic assay or have been previously studied via PubMed online search. |
|
| |
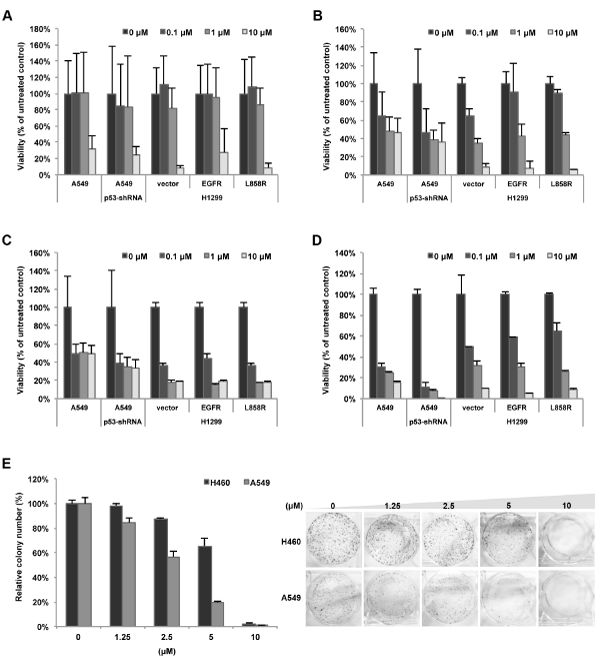
| Of the 84 small molecular drugs screened, 51 drugs had an effective cancer cell inhibitory effect of an IC50< 10 μM via MTT assay or with an effective 50% inhibition in a clonogenic assay at 10 μM. Among them, 35 drugs (69%) and 46 drugs (90%) were searched via PubMed online literature database and related to “lung cancer” and “cancer”, respectively. Figure 2A-D shows that four drugs (GW8510, tanespimycin, anisomycin, and pyrvinium) had cytotoxic effects on lung cancer cells and that there was no significant difference between the effects in these five cell lines. Additionally, the cytotoxic ability of either anisomycin or pyrvinium was higher than GW8510. The majority of cells were killed in the presence of 10 μM of GW8510, 1 μM of tanespimycin or 0.1 μM of either anisomycin or pyrvinium. Additional results are summarized in Table 2. Figure 2E shows the representative results of a clonogenic assay with A549 and H460 treated with chlorpromazine. Treatment with 10 μM chlorpromazine reduced the colonies over 50% in both A549 and H460. Twenty-three drugs reduced the clonogenic survival of these two lung cancer cells. In contrast, 33 drugs had no effect on cell inhibition in MTT or clonogenic assay, and are considered to not have cancer cell inhibition effects in vitro. However, through a PubMed medical literature database search of these 33 drugs, a small number of the drugs (e.g., procaine, apigenin, diltiazem, and nifedipine) were reported to have effects in different lung cancer or cancer cell lines (Supplemental Table S6). In this study, we did not include these drugs for further analysis. In summary, there are 62 effective drugs selected from the 95 potential drugs for NSCLC. |
| |
|
|
Figure 2: Cytotoxicity of 4 potential drugs. Drugs were tested in five lung cancer cell lines, A549, A14 (A549-p53 shRNA stable clone) and EGFR-expressing H1299 cells, parental (vector), wild-type EGFR (EGFR) and EGFR-L858R mutant (L858R). Cells were treated with GW8510 (A), tanespimycin (B), anisomycin (C), and pyrvinium (D) with various concentrations (0, 0.1, 1, 10 μM) for 72 hours, respectively. Cell viability was then evaluated using an MTT assay. (E) Lung cancer cells, both H460 and A549, were treated with chlorpromazine at 0, 1.25, 2.5, 5, 10 μM respectively for 10 days. Cell colonies were then counted and expressed in terms of percent colony number relative to the control. |
|
| |
| Evaluation the methods to query the CMap |
| |
| Based on this limited biochemical data along with PubMed search results (62 effective and 33 non-effective), we evaluated the accuracy of our analysis methods. First, we used the top 100 up/down probesets to select differential gene signatures for querying the CMap. The potential drugs from different early stage (stage I) and late stage (stage III/IV) NSCLC populations showed an accuracy rate of 73% in YM versus 72% in EAGLE and 62% in YM versus 61% in EAGLE, respectively. In addition, the accuracy of the predicted drugs in the unknown stage Blum dataset was 71%, suggesting that our predicted models are sensitive in different datasets (Supplemental Table S7A). Furthermore, an additional 4 methods were employed, and data fusion from the 5 methods to select input gene lists and perform accuracy rate analysis was performed. As shown in Supplemental Table S7, the accuracy rate from each method is generally less than 70%, for example, the top 1% ranked (59~67%), SAM (57~67%) and data fusion (58~66%), among the different lung cancer datasets. |
| |
| Treatment of trichostatin A results in tumor shrinkage and induces p21 expression |
| |
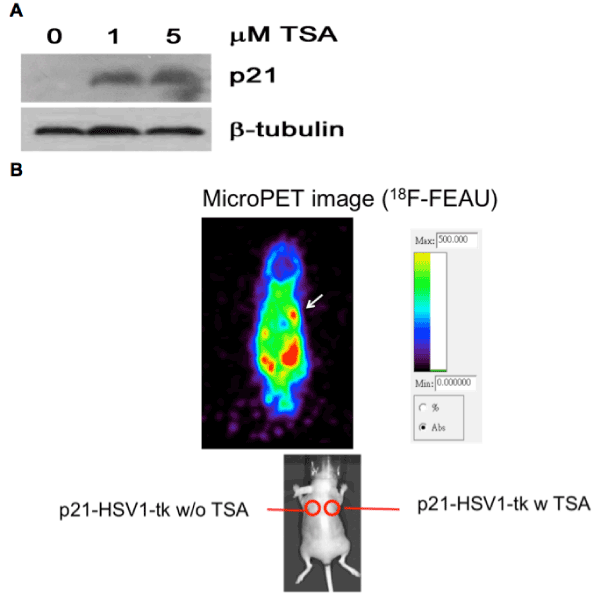
| Trichostatin A, a known HDAC inhibitor, induces transcription of cancer cells. Lung cancer cells have been studied regarding their hypermethylated status compared with normal lung cells, and many differentially expressed genes in NSCLC cells [22-25] after treatment with HDAC inhibitors have been reported. We noticed that p21 was significantly up-regulated in the CMap arrays after trichostatin A treatment and that p21 was down-regulated in NSCLC patient arrays. In fact, one of the mechanisms of trichostatin A induced cancer cell apoptosis is via up-regulation of p21 expression [26,27]. Consistent with previous observations, treatment of H1299 cells with trichostatin A resulted in the up-regulation of p21 (Figure 3A), and this drugtargeted interaction was confirmed via an in vivo animal model ( Figure 3B). These data and analyses suggest that the modulation of the expression of p21 and other similar genes might reveal drug-target relationships involved in NSCLC carcinogenesis. |
| |
|
|
Figure 3: Treatment of trichostatin A results in increase of p21 in vitro and in vivo. (A) H1299 cells were treated with trichostatin A for 0, 1 and 5 μM for 6 hours respectively. Protein expression level was then analyzed by Western blot with anti-p21 antibody. β-tubulin was used as an internal control. (B) In vivo non-invasive microPET imaging on the p21-HSV1-tk expressed H1299 mouse tumor model shows trichostatin A induced p21 overexpression (arrow) in the xenografted tumor. Enhanced accumulation of 18F-FEAU in the tumor is noted. The tumor without injection of trichostatin A on the left shoulder shows no enhanced uptake of radiotracer. |
|
| |
| Matrix visualization for gene expression profiling using a Generalized Association Plot (GAP) |
| |
| We next explored the relationships between drug-mediated signatures from the CMap and the signatures generated from our lung adenocarcinoma patients. Two final datasets were studied here for the 624 probesets (265 up-regulated and 359 down-regulated) selected for the 9 functional categories as in Table 1.The drug-level dataset has median of expression ratios for each of the 624 (probeset) by 62 (drug) combinations and the function-level dataset has median (of medians) of expression ratios for each of the 624 (probeset) by 9 (function) combinations. |
| |
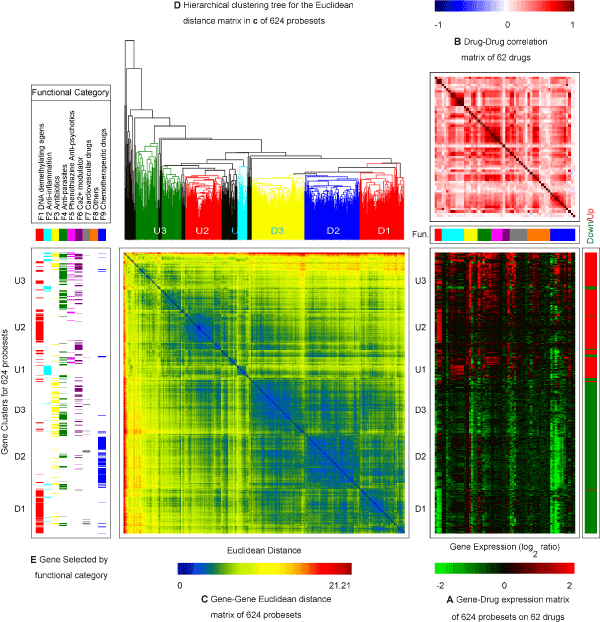
| Generalized association plots (GAP) [28-30] were used to illustrate (1) gene clusters; (2) drug (function) groups; and (3) interaction of gene clusters on drug groups for this expression in Figure 4. Three matrix maps are presented in this GAP display: a gene expression profile map of the 624 probesets by the 62 drugs (Figure 4A); a drugdrug correlation map between the 62 drugs (Figure 4B); and a genegene Euclidean distance map among the 624 probesets (Figure 4C). In addition to the three matrix map, a hierarchical clustering tree (Figure 4D) is applied to sort the Euclidean distance map in Figure 4C and a panel is used to illustrate gene selection mechanism by functional category (Figure 4E). To the right of Figure 4A is a covariate indicating if a probeset was selected as an up-(red) or down-(green) regulated gene. For Figure 4A & C, the 624 probesets were sorted by applying the HCT-R2E algorithm [29] in Figure 4D to the Euclidean distance map ( Figure 4C) for the 624 probesets. HCT-R2E is an improved hierarchical clustering tree (HCT) with the flipping of intermediate nodes guided by a global trend, which simultaneously identified coherent local clusters with smooth global patterns in given gene expression profiles. As can be observed with the up/down covariate, the 624 probesets were sorted into two main clusters of relatively up-regulated probesets (red) on the top portion and relatively down-regulated probesets (green) at the bottom part of Figure 4A. |
| |
|
|
Figure 4: Generalized association plots (GAP) for 624 probesets of the 62 drugs with the 9 functional categories. (A) The gene expression profile map of the 624 probesets by the 62 drugs; (B) The drug-drug correlation map between the 62 drugs; and (C) The gene-gene Euclidean distance map among the 624 probesets; (D) The hierarchical clustering tree for sorting the Euclidean distance map in (C); and (E) The panel for gene selection mechanism by functional category. To the right of Figure 4A is a covariate indicating if a probeset was selected as a up-(red) or down-(green) regulated gene. In (A) and (C), the 624 probesets were sorted using the HCT-R2E in (D). In (A) and (B) permutation of the 62 drugs were divided into two layers. HCT-R2E was used for both the ordering of functions at the outer layer and ordering of drugs within each inner layer for functions. |
|
| |
| The permutation of the 62 drugs in Figure 4A & B was divided into two layers, drug-layer and function-layer. The function-layer was used to determine order of the 9 functional categories using the functionlevel dataset while the drug-layer was employed to identify order of related drugs within each of the 9 functional groups using the drug-level dataset. For the function-layer, order of the 9 functional categories was determined by applying HCT-R2E to the Pearson correlation matrix among the 9 functions calculated using the function-level dataset. For drug-layer, HCT-R2E was also employed to sort, for each functional category, the among drug Pearson correlation matrix calculated using the drug-level dataset. |
| |
| From Figure 4B, we can visualize within function coherence of drugs on the main diagonal and between function drug correlation patterns on the off-diagonal. The functional categories of DNA demethylating agents, anti-inflammation, and phenothiazine anti-psychotics have more coherence (high positive correlation) within function relationships. Some sub-function structures that may be due to cell type or other mechanisms were observed as well. Most of the between function drugs have a weak positive correlation as denoted by lighter red (pink) colors on the off-diagonal. By cross-examining Figure 4A & C-E, we can roughly identify three relatively up-regulated (U1, U2, and U3) and three down-regulated (D1, D2, and D3) gene clusters among the 624 probesets: (D1) Genes mainly selected by DNA demethylating agents with a relatively strong decreased expression upon treatment with DNA demethylating agents and chemotherapeutic drugs; (D2) Genes selected by chemotherapeutic drugs only with a relatively decreased expression upon treatment with chemotherapeutic drugs only; (D3) Genes selected by antibiotics/anti-parasites/Ca2+modulator categories with a relatively low to medium down-regulation across all nine functional categories; (U1) A small group of genes screened by the anti-inflammation functional group with a strong up-regulation upon treatment with the anti-inflammation drugs and down-regulation upon treatment with the chemotherapeutic drugs; (U2) Up-regulated genes selected by DNA demethylating agents; (U3) Genes selected by anti-parasites/phenothiazine anti-psychotics/Ca2+modulator) categories with a relatively medium to high up-regulation across all nine functional categories. Several sub-gene clustering patterns were also determined using a more detailed classification. The most informative pieces of visual information are illustrated in Figure 4A for the interaction of gene clusters by functional group. |
| |
| To study the potential reversed gene expression signatures from the gene-function (drug) profiling of the CMap database to the gene-patient profiling of our patients, Supplemental Figure S1 was constructed. Supplemental Figure S1 has the expression profiles (log2 ratio of tumor to adjacent normal) of the same set of 624 probesets from Figure 4A on the 44 patients. In the middle of the two panels are the covariates indicating the up- and down-regulated patterns from Figure 4A and corresponding to the up- and down-regulated patterns derived from the 44 patients. Among the 624 probesets, 514 fell into the category of non-differentially expressed genes for the 44 patient profiles. These 514 probesets were not selected by any of the 44 patients as differentially expressed genes (neither up nor down). Among the remaining 110 differentially expressed probesets, 89 were considering reversed signature probesets. Of these, 45 (12.5%) followed the upregulated (patient) to down-regulated (CMap) pattern, whereas the remaining 44 (16.6%) probesets followed the reversed down-regulated (patient) to up-regulated (CMap) pattern (Supplemental Table S8A & B). |
| |
| The 45 up- and 44 down-regulated signatures in NSCLC patients, which were reversed by 9 functional drug categories, were analyzed via the ConsensusPathDB-human pathway analyzer [31] to identify and determine the most significant pathways of the up- and down-regulated with p-value and q-value, respectively. There were 8 up-regulated and 6 down-regulated significant pathways in NSCLC patients, including the SCF(Skp2)-mediated degradation of p27/p21 (CCNA2, CCNE1 and CCNE2), the p53 signaling pathway (CCND1, CCNE1, and CCNE2), and the direct p53 effectors (ATF3, CDKN1A, DUSP1, HSPA1A, and LIF). Because p53 mutation has been observed in most NSCLC patients, such reverse relationships could further highlight the possibility of targeting p53 associated pathways [32]. Importantly, we identified that CDKN1A (p21) was reversed in the down-regulated signatures of NSCLC patients to the up-regulated signatures of the CMap (Supplemental Table S8B). Moreover, p21 appeared in the “direct p53 effector” pathway that is an important pathway in relation to cancer and p53 is the most frequently mutated gene in lung adenocarcinoma [33]. |
| |
| Discussion |
| |
| In this study, we used five different gene selection methods to generate a list of 363 potential drugs (Figure 1), which are commonly occurred in the different studied populations. Additionally, a total of 62 in vitro validated drugs were categorized into 9 functional groups with anti-cancer effects in NSCLC, and gene expressions were correlated with each function. Finally, we obtained the reversal gene signatures from potential drugs to patient samples. This study may aid in the understanding of the carcinogenesis of NSCLC as well as the interactions between tumor, drug, and patient. |
| |
| In addition to display the cytotoxicity of different functional drugs, the representative results of the in vitro cytotoxic and clonogenic tests for five drugs (tanespimycin, GW-8510, anisomycin, pyrvinium, and chlorpromazine) are shown as Figure 2. These drugs were from the different functional groups categorized in Table 2 (anti-inflammation, chemotherapeutic drugs, antibiotics, antiparasitic, and phenothiazine anti-psychotics respectively) and individually showed effective results (IC50 < 10 μM) independent of p53 or EGFR expression. These results imply that these drugs might overcome the mutation of p53 or EGFR, which are both frequently found in the NSCLC patients. Consistent with previous studies, tanespimycin, GW-8510, and anisomycin have been shown to exhibit effective cytotoxicity by targeting X-linked inhibitor of apoptosis (XIAP), EGFR signaling, and c-jun NH2-terminal kinase pathways in p53 mutant or EGFR wild type/TKI resistant NSCLC cells, respectively [34-36]. However, the downstream signaling of pyrvinium is unclear and deserves further study. |
| |
| The study by Wang et al. [37] and this study both used the same published microarray data (GSE7670 and GSE10072). Wang et al. [ 37] used a two-sample t-test to identify differential genes from these two datasets, which generated 434 differential probesets (125 up and 309 down) and 530 differential probesets (180 up and 350 down), respectively. They further intersected these two sets of differential genes to produce a common gene signature (93 up and 250 down). In contrast, we used each patient’s signature to identify differential genes. Moreover, a common drug may not be suitable for all patients who have the same disease clinically. To improve the degree of credibility, we enlarged the dataset by generating another adenocarcinoma lung cancer dataset (GSE27262) and used the Blum dataset to validate the results. Comparing the differential genes between Wang’s study and our top 100 up/down results, all 93 up-regulated genes and 232 of the 250 down-regulated genes from the previous study are involved in our upregulated list and down-regulated list, respectively. In particular, some genes had a high frequency in our list; for example, TOX3 (previously known as TNRC9) appeared in the up-regulated gene list of 45 patients, and AGER (RAGE) appeared in the down-regulated gene list of 66 patients but not included in Wang’s list (Supplemental Table S9A & B). TOX3 is associated with breast cancer for BRCA1/BRCA2 mutation carriers and may be a strong carcinogenetic gene of cancer [38-40]. The expression for RAGE is significantly reduced in lung adenocarcinoma cancer tissue compared with adjacent normal lung tissue, supporting the view that RAGE is a tumor suppressor gene in lung cancer [41- 43]. Conversely, KLF4 (221841_s_at) was down-regulated in patients, but was up-regulated in the CMap. Moreover, KLF4 showed a high frequency in our gene signature list but was not included in Wang’s list [37]. Additionally, of the reversed probesets, only one up-regulated probeset and 12 down-regulated probesets from the patient signatures were included in Wang’s 93 up-regulated and 250 down-regulated probesets list, respectively. Therefore, our approach could retrieve more reverse genes for analysis than Wang’s study and might represent the most important target gene of lung cancer patients. Finally, several probesets representing the same gene were revealed concurrently in our analysis, such as TOP2A, COL10A1, COL11A1 or COL1A1 among up-regulated (Supplemental Table S9A) and SFTPC or ADH1B among down-regulated (Supplemental Table S9B) genes, which support the confidence of our analysis. |
| |
| As summarized in Supplemental Table S10, there have been many reports using the CMap tool to target specific signaling pathways. Here, we demonstrate that certain groups of drugs can provide a reverse signature of carcinogenesis and that the embedded signatures can be revealed in the patient samples (Supplemental Figure S1). This approach provides further insight into the complex carcinogenic pathways in NSCLC. We have successfully identified cancer cell apoptosis in the presence of p21 overexpression after HDAC inhibitor treatment, both in vitro and in vivo. By using this platform, other pathways or associated genes of interest can be evaluated more efficiently with their previous unknown or unclear functions. |
| |
| EGFR TKIs, such as gefitinib or erlotinib, were not identified from the CMap analysis, especially in the YM dataset, which included many EGFR mutant patients and may have high response to EGFR-TKIs. The possible explanation is that there is no chip for erlotinib treatment and only one chip with gefitinib treatment in the CMap array dataset. Other common chemotherapy drugs for NSCLC patients, such as cisplatin, pemetrexed, or taxanes also cannot be revealed due to lack of data in the CMap. However, while performing PubMed search for the 62 validated drugs in the clinical trial registration website: https://clinicaltrials.gov/, several drugs have undergone clinical trials, especially in the DNA demethylating agents (vorinostat and azacitidine) and anti-inflammation (tanespimycin, acetylsalicylic acid, and sirolimus) functional groups. Several drugs (resveratrol, alvespimycin, verteporfin, and trioxsalen) under recruiting status of clinical trials suggested current clinical development. The potential for other functional categories without current clinical trials warrant further studies. In conclusion, the study result categorized nine functional groups of anti-cancer drugs and repurposed most old drugs to new anti-cancer effects, which will be helpful in the development of new cancer treatment strategy. |
| |
| |
| References |
| |
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, et al. (2008) Cancer statistics, 2008. CA Cancer J Clin58: 71-96.
- Department of Health tEY (2007) Cancer registry annual report in Taiwan area, Department of Health, Executive Yuan, ROC.
- Thatcher N, Chang A, Parikh P, Rodrigues Pereira J, et al. (2005) Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet366: 1527-1537.
- Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, et al. (2005) Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med353: 123-132.
- Huang SF, Liu HP, Li LH, Ku YC, Fu YN, et al. (2004) High frequency of epidermal growth factor receptor mutations with complex patterns in non-small cell lung cancers related to gefitinib responsiveness in Taiwan. Clin Cancer Res10: 8195-8203.
- Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, et al. (2005) Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst97: 339-346.
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, et al. (2006) The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science313: 1929-1935.
- Lamb J (2007) The Connectivity Map: a new tool for biomedical research. Nat Rev Cancer7: 54-60.
- Hassane DC, Guzman ML, Corbett C, Li X, Abboud R, Young F, et al. (2008) Discovery of agents that eradicate leukemia stem cells using an in silico screen of public gene expression data. Blood111: 5654-5662.
- Rosenbluth JM, Mays DJ, Pino MF, Tang LJ, Pietenpol JA (2008) A gene signature-based approach identifies mTOR as a regulator of p73. Mol Cell Biol28: 5951-5964.
- Setlur SR, Mertz KD, Hoshida Y, Demichelis F, Lupien M, et al. (2008) Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst100: 815-825.
- Su LJ, Chang CW, Wu YC, Chen KC, Lin CJ, et al. (2007) Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics8: 140.
- Landi MT, Dracheva T, Rotunno M, Figueroa JD, Liu H, et al. (2008) Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS One3: e1651.
- Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A98: 5116-5121.
- Wu CC, Yu CT, Chang GC, Lai JM, Hsu SL (2011) Aurora-A promotes gefitinib resistance via a NF-?B signaling pathway in p53 knockdown lung cancer cells. Biochem Biophys Res Commun405: 168-172.
- Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, et al. (1997) Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am J Respir Cell Mol Biol17: 353-360.
- Chen YR, Fu YN, Lin CH, Yang ST, Hu SF, et al. (2006) Distinctive activation patterns in constitutively active and gefitinib-sensitive EGFR mutants. Oncogene25: 1205-1215.
- Liu RS, Hsieh YJ, Ke CC, Chen FD, Hwu L, et al. (2009) Specific activation of sodium iodide symporter gene in hepatoma using alpha-fetoprotein promoter combined with hepatitis B virus enhancer (EIIAPA). Anticancer Res29: 211-221.
- Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, et al. (2007) A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med356: 11-20.
- Lau SK, Boutros PC, Pintilie M, Blackhall FH, Zhu CQ, et al. (2007) Three-gene prognostic classifier for early-stage non small-cell lung cancer. J Clin Oncol25: 5562-5569.
- Skrzypski M, Jassem E, Taron M, Sanchez JJ, Mendez P, et al. (2008) Three-gene expression signature predicts survival in early-stage squamous cell carcinoma of the lung. Clin Cancer Res14: 4794-4799.
- Tang SC, Wu MF, Wong RH, Liu YF, Tang LC, et al. (2011) Epigenetic mechanisms for silencing glutathione S-transferase m2 expression by hypermethylated specificity protein 1 binding in lung cancer. Cancer117: 3209-3221.
- Richards KL, Zhang B, Sun M, Dong W, Churchill J, et al. (2011) Methylation of the candidate biomarker TCF21 is very frequent across a spectrum of early-stage nonsmall cell lung cancers. Cancer117: 606-617.
- Chen M, Voeller D, Marquez VE, Kaye FJ, Steeg PS, et al. (2010) Enhanced growth inhibition by combined DNA methylation/HDAC inhibitors in lung tumor cells with silenced CDKN2A. Int J Oncol37: 963-971.
- Ibanez de Caceres I, Cortes-Sempere M, Moratilla C, Machado-Pinilla R, Rodriguez-Fanjul V, et al. (2010) IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene29: 1681-1690.
- Zhang F, Zhang T, Teng ZH, Zhang R, Wang JB, et al. (2009) Sensitization to gamma-irradiation-induced cell cycle arrest and apoptosis by the histone deacetylase inhibitor trichostatin A in non-small cell lung cancer (NSCLC) cells. Cancer Biol Ther8: 823-831.
- Mukhopadhyay NK, Weisberg E, Gilchrist D, Bueno R, Sugarbaker DJ, et al. (2006) Effectiveness of trichostatin A as a potential candidate for anticancer therapy in non-small-cell lung cancer. Ann Thorac Surg81: 1034-1042.
- Chen Ch (2002) Generalized association plots: Information visualization via iteratively generated correlation matrices. Statistica Sinica12: 7-29.
- Tien YJ, Lee YS, Wu HM, Chen CH (2008) Methods for simultaneously identifying coherent local clusters with smooth global patterns in gene expression profiles. BMC Bioinformatics9: 155.
- Wu HM, Tien YJ, Chen Ch (2010) GAP: A graphical environment for matrix visualization and cluster analysis. Comput Stat Data Anal54: 767-778.
- Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, et al. (2011) ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res39: D712-D717.
- Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, et al. (2010) Diverse somatic mutation patterns and pathway alterations in human cancers. Nature466: 869-873.
- Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, et al. (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature455: 1069-1075.
- Dong F, Guo W, Zhang L, Wu S, Teraishi F, et al. (2006) Downregulation of XIAP and induction of apoptosis by the synthetic cyclin-dependent kinase inhibitor GW8510 in non-small cell lung cancer cells. Cancer Biol Ther5: 165-170.
- Sawai A, Chandarlapaty S, Greulich H, Gonen M, Ye Q, et al. (2008) Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res68: 589-596.
- Abayasiriwardana KS, Barbone D, Kim KU, Vivo C, Lee KK, et al. (2007) Malignant mesothelioma cells are rapidly sensitized to TRAIL-induced apoptosis by low-dose anisomycin via Bim. Mol Cancer Ther6: 2766-2776.
- Wang G, Ye Y, Yang X, Liao H, Zhao C, et al. (2011) Expression-based in silico screening of candidate therapeutic compounds for lung adenocarcinoma. PLoS One6: e14573.
- Ripperger T, Gadzicki D, Meindl A, Schlegelberger B (2009) Breast cancer susceptibility: current knowledge and implications for genetic counselling. Eur J Hum Genet17: 722-731.
- Antoniou AC, Sinilnikova OM, McGuffog L, Healey S, Nevanlinna H, et al. (2009) Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Hum Mol Genet18: 4442-4456.
- Antoniou AC, Beesley J, McGuffog L, Sinilnikova OM, Healey S, et al. (2010) Common breast cancer susceptibility alleles and the risk of breast cancer for BRCA1 and BRCA2 mutation carriers: implications for risk prediction. Cancer Res70: 9742-9754.
- Rehbein G, Simm A, Hofmann HS, Silber RE, Bartling B (2008) Molecular regulation of S100P in human lung adenocarcinomas. Int J Mol Med22: 69-77.
- Rho JH, Roehrl MH, Wang JY (2009) Glycoproteomic analysis of human lung adenocarcinomas using glycoarrays and tandem mass spectrometry: differential expression and glycosylation patterns of vimentin and fetuin A isoforms. Protein J28: 148-160.
- Jing R, Cui M, Wang J, Wang H (2010) Receptor for advanced glycation end products (RAGE) soluble form (sRAGE): a new biomarker for lung cancer. Neoplasma57: 55-61.
|
| |
| |