| Research Article |
Open Access |
|
| Hsin-I Chang1, Ming-Yen Cheng1 and Ming-Kung Yeh2* |
| 1Department of Biochemical Science and Technology, National Chia Yi University, Taiwan, Republic of China |
| 2Institute of Preventive Medicine National Defence Medical Center, Taipei, Taiwan, Republic of China |
| *Corresponding authors: |
Ming-Kung Yeh
Institute of Preventive Medicine National Defence Medical Center
161, Sec. 6, Ming-chung R.D, Taipei
(11490), Taiwan, Republic of China
Tel: +886-5-2717923
Fax: +886-5-2717780
E-mail: hchang@mail.ncyu.edu.tw |
|
| |
| Received October 04, 2011; Published July 27, 2012 |
| |
| Citation: Chang HI, Cheng MY, Yeh MK (2012) Clinically-Proven Liposome-Based Drug Delivery: Formulation, Characterization and Therapeutic Efficacy. 1: 195. doi:10.4172/scientificreports.195 |
| |
| Copyright: © 2012 Chang HI, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| Liposomes in their various forms have the possibility of providing therapeutic efficacy in the area of drug delivery. This review mainly addresses therapeutic effect of current clinical approved liposomal formulations, preparation techniques, storage aspects, as well as lipid compositions. Researches on liposome formulations have progressed from conventional vesicles to new generation liposomes, such as cationic liposomes, temperature-sensitive liposomes and virosomes, by modulating the preparation method and lipid composition. Furthermore, the major preclinical and clinical data relating the principal liposomal formulations are also summarized. |
| |
| Keywords |
| |
| Liposomes; Temperature-sensitive liposomes; Lyophilization; Virosomes |
| |
| Introduction |
| |
| The clinical utility of most conventional chemotherapeutics is limited either by the inability to deliver therapeutic drug concentrations to the target tissues or by severe and harmful toxic effects on normal organs and tissues. Controlled drug delivery systems have been attempted to overcome these problems by providing selective delivery to the affected area. Liposomes are small, spherical and enclosed compartments separating an aqueous medium from another by phospholipid bilayer and liposomal formulations are one of advanced drug delivery systems in clinical application. Due to differences in preparation methods and lipid compositions, liposomes can be classified according to their lamellarity (uni- and multi-lamellar vesicles), size (small, intermediate, or large) and charge (anionic, cationic and neutral) [1-3]. Liposomes are able to encapsulate lipophilic or hydrophilic drugs with their lipidic layers or in their aqueous core respectively and deliver those to target site for in vivo application. Moreover, liposome delivery system can increase the solubility of hydrophobic drugs and stabilize a variety of therapeutic agents such as peptides, proteins and nucleotides in bloodstream [3,4]. In clinic studies, liposomes show improved pharmacokinetics and biodistribution of therapeutic agents and thus minimize toxicity by their accumulation at the target tissue [5]. Liposomes were first discovered by Bangham in 1965 and the first liposomal pharmaceutical product, Doxil, received FDA approval in 1997 for the treatment of chemotherapy refractory AIDS-related Kaposi’s sarcoma [4,5]. Currently there are about 12 liposome-based drugs approved for clinical use and more are in various stages of clinical trials (Table 1 and 2) [5-46]. Most of liposomal drug formulations, including Ambisome, Doxil and Myocet, are approved for intravenous application. Other administration routes such as intramuscular delivery have also been approved for delivery of surface antigens derived from the hepatitis A or influenza virus. Oral delivery has been examined however this is more troublesome due to the potential for liposome breakdown following exposure to bile salts [47]. |
| |
|
|
Table 1: Liposome based drugs in market. |
|
| |
|
|
Table 2: Liposome based drug in clinical trials. |
|
| |
| Liposomal encapsulation technology |
| |
| Many hundreds of drugs, including anti-cancer and antimicrobial agents, chelating agents, peptide hormones, enzymes, other proteins, vaccines and genetic materials, have been incorporated into the aqueous or lipid phases of liposomes with various sizes, compositions and other characteristics by different preparation techniques. An ideal method of liposome formulation is preparing liposome with high entrapment efficiency, narrow particle size distribution and long term stability. Numbers of techniques have been reported for preparation of liposomes such as Bangham method, the detergent depletion method, the ether/ethanol injection method, the reverse phase evaporation and the emulsion method [48]. The majority of liposome preparation methods require using organic solvents to dissolve lipids but these organic solvents are harmful to the environment and human body. Recently, some alternative methods including dense gas and supercritical fluid techniques have been introduced for liposome preparation without using any organic solvent [48-50]. Despite the clear advantages of dense gas or supercritical fluid liposome production, there are also problems with the known processes for liposome formation. The dense gas or supercritical fluid processes generally require elevated pressures of at least 1,000 psi and the conditions commonly used are 3000–4500 psi and temperatures of 60°C for liposome production. For drug encapsulation, both antisolvent methods successfully encapsulated low molecular weight drugs such as paclitaxel, camptothecin and betulinic acid in liposomes [50]. However, high temperature, pressure, and shearing forces presenting in liposome processing condition with dense gas or supercritical fluid potentially denature high molecular weight drugs, such as peptides or proteins and result low production and encapsulation efficiency [51]. Physicochemical properties of liposomal formulations, including size, membrane lamellarity, surface charge, permeability, and encapsulation volume, are depending on the lipid composition (cationic, anionic, and neutral lipid species). The major function of liposome preparation techniques is to obtain efficient drug entrapment and increase stability of the liposome products [52]. Most of drugs listed in Table 1 using liposomes as carrier to increase the drug solubility in aqueous solution and also decrease drug toxicity in human body. Ambisome, Doxil and Myocet are the examples for improving therapeutic index by reducing the toxicities associated with the free drugs. |
| |
| Storage of Liposomes: lyophilization |
| |
| Liposomes dispersed in aqueous solution generally face physical and chemical instabilities after long term storage [53]. Hydrolysis and oxidation of phospholipids and liposome aggregation are the common cause of liposome instabilities. According to the literature, many methods have been investigated for the stabilization of liposomes, such as lyophilization, freezing and spraying drying. In commercial liposome-based drugs (Table 1), AmBisome, Amphotec, Myocet, Visudyne and LEP-ETU are all lyophilized products. In general, freeze-drying increases the shelf-life of liposomal formulations and preserves it in dried form as a lyophilized cake to be reconstituted with water for injection prior to administration [54]. Furthermore, cryoprotectants need to be added to maintain particle size distribution of liposomes after freeze-drying- rehydration cycle. Various types and concentrations of sugars have been investigated for their ability to protect liposomes against fusion and leakage during lyophilization processes [54]. In commercial liposome lyophilized products, lactose was used as a cryoprotectant in the formulation of Amphotec, Myocet and Visudyne and sucrose was added in the formulation of Ambisome and LEP-ETU to increase liposome stability during lyophilization. |
| |
| Interestingly, these commercial lyophilized products showed similar shelf-life in comparison with other liposome products (eg: suspension and emulsions) and hence lyophilization may not have expected effect on liposome stability. In 1998, Clemons et al. [55] compared the potency and therapeutic efficacy among the different lipid-based formulations of amphotericin B (Amphotec, AmBisome and Abelcet) for the treatment of systemic and meningeal cryptococcal disease. Their work indicated that the therapeutic efficacy of Amphotec and AmBisome was superior to that of Abelcet by up to 10-fold in survival and in clearing infection from all organs. In these three commercially available lipid-based formulations of amphotericin B, Amphotec and AmBisome are both lyophilized products and Abelcet is formulated as a suspension form. Therefore, lyophilization may not extend the shelflife of products but may increase therapeutic efficacy in vivo. Similar results were also reported in our previous studies. We investigated the stability of the siRNA-loaded liposomes in suspension and lyophilized powder form up to 1 month post manufacture [56]. Following formulation, the siRNA-loaded liposomes were stored at either 4°C or room temperature. The particle size and zeta potential of siRNAloaded liposomes remained unchanged for both storage conditions. However, siRNA entrapment efficiencies for both storage conditions were observed to have decreased slightly over time. Surprisingly, the gene-silencing efficiency of siRNA-loaded liposomes in aqueous solution was almost completely abolished following 1-month of storage at either 4°C or room temperature. This was in contrast to liposomes prepared in the lyophilized powder form where 100% of the genesilencing efficiency was retained following storage at either 4°C or room temperature for a month. Although therapeutic efficiency of liposomebased drug may vary depending on the choice of lipids, the preparation technique, the physico-chemical characteristics of the bioactive, and the overall charge of the liposome, lyophilisation is absolutely essential for the long term storage of liposome-based drugs. |
| |
| Liposomal anti-cancer drug researches: Doxorubicin |
| |
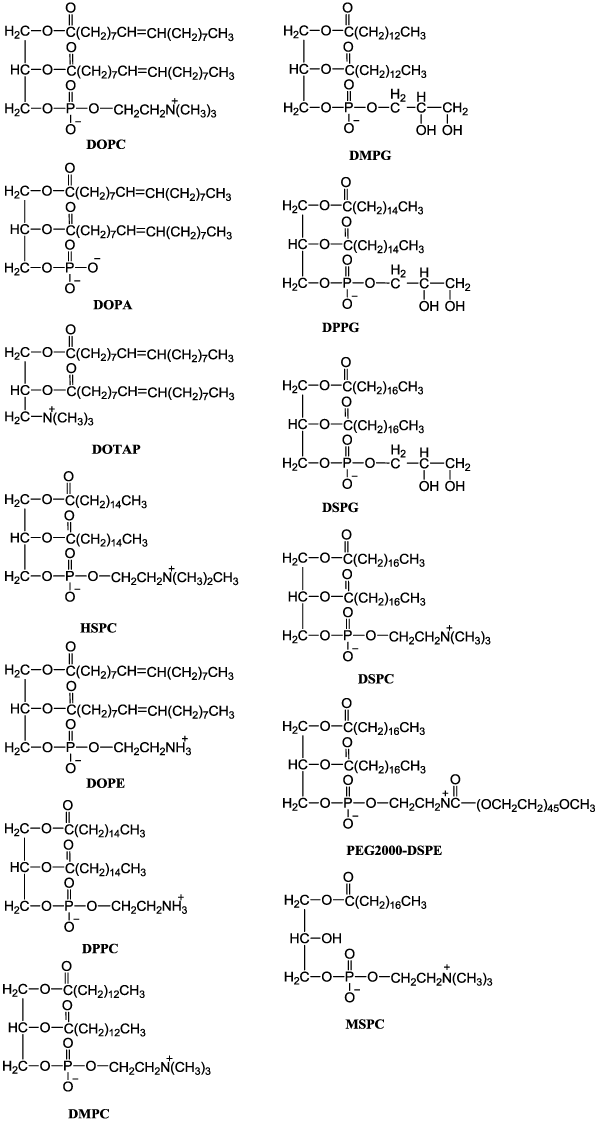
| Liposome delivery systems offer the potential to enhance the therapeutic index of anticancer drugs, either by increasing the drug concentration in tumor cells and by decreasing the exposure in normal host tissues. Doxorubicin is an anthracycline widely used to treat solid and hematological tumors, but its major drawback is the onset of resistance. Therefore, doxorubicin-loaded liposomes were developed to combat aggressive tumors, like breast and ovary metastatic cancers and Kaposi’s sarcoma. Myocet and Doxil were first approved liposomebased drugs for cancer treatment. Both products contain doxorubicin but different particularly in the presence of Poly Ethylene Glycol (PEG) coating (Figure 1). In pharmacokinetic studies of doxorubicin-loaded liposomes, free doxorubicin had an elimination half-life time of 0.2 hours and an AUC (area under the cure) of 4 μg h ml-1 in patients an as compared with 2.5 hour and 45 μg h ml-1 for Myocet and with 55 hours and 900 μg h ml-1 for Doxil, respectively [15]. Both liposome products showed longer circulating half-life as compared with free drug. In phase III head to head comparison of free doxorubicin vs Myocet in patients with metastatic breast cancer, similar results presented in response rates (26% for both) and progression-free survival times (4 months for both) but Myocet had low incidence of cardiac events (29 vs 13%) and of congestive heart failure (8 vs 2%) [57]. Therefore, Myocet tends to reduce drug-related toxicity (eg: cardiotoxicity) rather than to enhance antitumor efficacy. Similar to Myocet, Doxil had a better safety profile including the reduce of cardiotoxicity, myelosuppression, vomiting and alopecia in phase III trial of metastatic breast cancer whereas its response rates, progression-free survival times and overall survival times demonstrated equivalent efficacy to conventional doxorubicin. |
| |
|
|
Figure 1: Chemical structures of lipids in liposome formulations. |
|
| |
| Lipo-dox is the second generation of PEGylated liposomal doxorubicin and which is composed of Distearoyl Phosphatidycholine (DSPC) and cholesterol with surface coating with PEG [17]. Liposomes composed of phospholipids like DSPC (Figure 1), which has two completely saturated fatty acids (stearic acid), have higher stability compared with others containing unsaturated fatty acid (egg PC) or fatty acids of shorter or not uniform carbon chains like Hydrogenated Soy Phosphatidycholine (HSPC). In phase I clinical study, Lipo-dox has achieved the most prolonged circulation half-life (65 hours). However, the antitumor activity of Lipo-dox for hepatocellular carcinoma is not higher than free doxorubicin. Moreover, stomatitis became the new dose-limiting toxicity of PEGylated liposomal doxorubicin. For Lipodox, stomatitis appeared at doses of 30mg/m2 and reached dose limit at 50mg/m2. In contrast, Doxil reached dose limit at 80mg/m2 and hence Lipo-dox had higher incidence of serve stomatitis than Doxil. PEGylated liposomal doxorubicin, Doxil and Lipodox (both PEGylated form of liposomal doxorubicin) have significantly more side effects than Myocet (the non PEGylated form of liposomal doxorubicin) and this is mainly due to the long circulation properties of PEGylated liposomes. |
| |
| The new generation of doxorubicin-loaded liposomes is Thermosensitive Liposomes (TSL) which releases their encapsulated drugs in regions where local tissue temperatures are elevated. Compared with non-TSLs that remain stable and do not release drug in the physiologic temperature range, TSLs undergo a gel-to-liquid crystalline phase change when heated that renders the liposomes more permeable, releasing their encapsulated drugs. ThermoDox®, a proprietary TSL encapsulation of doxorubicin, recently is in phase III clinical trials for the treatment of hepatocellular carcinoma. ThermoDox® is composed of Dipalmitoylphosphatidylcholine (DPPC), Monostearoylphosphatidylcholine (MSPC) and PEG 2000-DSPE in 90:10:4 molar ratio [35-36]. In the design of TSL, it is necessary to choose a phospholipid that has a gel-to-liquid crystalline phase transition temperature (Tc) in the temperature range of clinically attainable local hyperthermia (41- 42°C). The mechanism behind TSL is the temperature induced membrane instability at the Tc of the used lipids. DPPC with a Tc=41.5°C, is an ideal lipid according to temperature triggered technology [58]. For liposomes composed of DPPC alone, the rate of release and the amount released are relatively small. By incorporating a small amount of lysolipids, such as MSPC or Monopalmitoylphosphatidylcholine (MPPC), into DPPC liposomes, Tc is shifted down slightly and membrane instability and drug release rate is significantly enhanced at Tc. Banno et al. [59] demonstrated that the presence of MSPC, rather than DSPE-PEG2000, in DPPC liposomes would give rise to the rapid drug release profile in vitro and that represents lysolipid is the more important component in determining TSL contents release. Indeed, Banno’s in vivo data showed that the presence of 9.6 mol% MSPC in TSL could result in more rapid elimination of the encapsulated doxorubicin (T1/2=1.29h), compared to the formulation without lysolipid (T1/2=2.91h). In 2007, Dromi et al. [36] compared the accumulation of doxorubicin in mice tumors among free doxorubicin, Doxil and ThermoDox. Results showed that over time, doxorubicin gradually increased in tumors when both Doxil and ThermoDox were used but not with free doxorubicin. At 24 hours after administration, doxorubicin concentrations in tumors were found to be significantly higher with Doxil than ThermoDox. ThermoDox is currently under evaluation in clinical trials and hence the therapeutic efficacy of ThermoDox is still unknown. |
| |
| Liposomal anti-cancer drug researches: daunorubicin and paclitaxel |
| |
| Daunorubicin and paclitaxel have also incorporated into liposomes for the formulation of liposomal anti-cancer chemotherapy drugs. DaunoXome is a commercial liposomal formulation of daunorubicin in which the drug is entrapped into small unilamellar vesicles composed of Distearoyl phosphatidylcholine (DSPC) and cholesterol in 2:1 molar ratio. LEP-ETU and EndoTAG-1 (previously called MBT-0206) are the potential liposomal formulations of paclitaxel and both are in clinical trials (Table 1 and 2). In comparison with conventional daunorubicin, DaunoXome was 36-fold higher in AUC and in vivo experiments indicated increased uptake of daunoXome in tumour tissue at 24 h. In phase III trial of DaunoXome versus vincristine in AIDS-related Kaposi's sarcoma, the efficacy of DaunoXome was comparable to that of vincristine. Response rates (25% vs 28%), time to treatment failure (115 vs 99 days), and overall survival (369 vs 342 days) were similar on both treatment arms and hence DaunoXome may provide another safe and effective chemotherapy [60]. |
| |
| Taxol® (paclitaxel) is a marketed product for the treatment of ovarian, breast, non-small cell lung cancer and AIDS-related Kaposi's Sarcoma [27]. It is one of the most effective anticancer drugs available on the market. However, paclitaxel is only sparingly soluble in water and therefore, intravenous administration depends on the use of the nonionic surfactant Cremophor EL (polyethoxylated castor oil) to achieve a clinically relevant concentrated solution. Unfortunately, Cremophor EL increases toxicity and leads to hypersensitivity reactions in certain patients. LEP-ETU formulation of paclitaxel is being developed to potentially reduce toxicities associated with Taxol®, by eliminating the drug formulation component polyoxyethylated castor oil. LEP-ETU formulations composed of 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), cholesterol and cardiolipin in 90:5:5 molar ratio were prepared by the modified thin-film hydration method. DOPC, a zwitter ionic natural phospholipid, is first chosen as one of the lipid components in LEP-ETU formulation because of a low Tc (−22°C), and which forms more flexible liposomes to entrap highly hydrophobic molecules. Moreover, cholesterol is included in LEP-ETU formulations to increase the liposome stability. In cardiotoxicity, positively charged doxorubicin's affinity for negatively charged cardiolipin, a lipid abundant in heart tissue, is thought to be involved in drug localization in the heart tissue [61]. Liposomes containing cardiolipin, reportedly reduced cardiotoxicity associated with doxorubicin by altering the pharmacokinetics and tissue distribution of the drug and hence cardiolipin may also exert similar results in LEP-ETU. Fetterly et al. [62,63] evaluated the Maximum Tolerated Dose (MTD), Dose-Limiting Toxicities (DLT), and pharmacokinetics of Liposome-Encapsulated Paclitaxel (LEP-ETU) in comparison to Taxol®. The MTD of LEPETU was 325 mg/m2 in phase I study of patients with locally advanced or metastatic carcinoma. This dose is higher than that achieved with Taxol, which is typically delivered at a dose range of 135 to 200 mg/ m2. The major toxicity to administration of paclitaxel is neuropathy. In the phase I study, neurotoxicity occurred in 5 of 12 patients (42%) treated with LEP-ETU at ≥325 mg/m2. Although a direct comparison with Taxol® is not possible, neutropenia was seen in 53% of metastatic breast cancer patients treated with 250 mg/m2 Taxol® as demonstrated by Winer et al. [64]. Therefore, the neuropathy caused by LEP-ETU appears to be no worse than that reported for Taxol® within 3 weeks of treatment. Following LEP-ETU administration, paclitaxel blood concentrations declined polyexponentially and AUC of paclitaxel was less than proportional with increasing dose, which is similar to Taxol®. Although similarities exist between the plasma pharmacokinetics of the two formulations, the clinical evidence obtained from the Phase I study shows LEP-ETU can be administered safely at higher doses than Taxol®. |
| |
| Another liposome formation of paclitaxel is Endo TAG-1 [28- 30]. The formulation of Endo TAG-1 is prepared by 1,2-Dioleoyl- 3-Trimethylammonium Propane (DOTAP), DOPC and paclitaxel in 50:47:3 molar ratio. DOTAP is a cationic synthetic lipid, which comprises one positive charge at the head group. The use of cationic lipids to enhance gene delivery has been studied extensively, but their application in clinic is relatively unexplored. Recently, there has been great interest in cationic liposomes, mainly due to their inherent ability to selectively target tumor vasculature. This selective affinity of cationic liposomes to tumor vasculature provides an opportunity for the development of many anti-angiogenic and anticancer formulations based on cationic liposomes [28]. Endo TAG-1 is the first formulation of cationic liposomes carrying paclitaxel in clinical trial. Endo TAG-1, which is currently tested in clinical studies, comprises about 3 mol% paclitaxel in a DOTAP/DOPC lipid matrix. For commercial storage, Endo TAG-1 formulations are lyophilized, and they are reconstituted with water for injection directly prior use. In preclinical programs, EndoTAG1-1 inhibited tumor growth also in Taxol-resistant animal tumor models, as for example, B16 melanoma and Sk-Mel 28 melanoma. EndoTAG1-1 demonstrated a strong antivascular effect on the preexisting tumor vasculature and affected several tumor microcirculatory parameters. In a Phase II trial of patients with pancreatic adenocarcinoma who were not candidates for surgery, EndoTag-1 in combination with gemcitabine substantially extended overall survival compared with gemcitabine alone [65]. Median survival in patients who received gemcitabine alone was 7.2 months, whereas it was up to 9.4 months in those who received combination treatment of EndoTAG-1 plus gemcitabine. After 6 and 12 months of treatment, survival rate was superior for all EndoTAG-1 doses plus gemcitabine compared with gemcitabine alone. The 12-month survival rates in patients given the two higher doses of EndoTAG-1 (22 and 44 mg/m2 plus gemcitabine) were 36% and 33%, respectively, compared with 17.5% in those given gemcitabine alone. Combination treatment with EndoTAG-1 plus gemcitabine was well tolerated and led to substantially prolonged survival rates compared to standard therapy in this phase II trial. Further clinical studies are warranted to demonstrate a statistically significant survival benefit associated with EndoTAG-1 plus gemcitabine in advanced pancreatic cancer. |
| |
| Liposome application in vaccine formulation: Epaxal and Inflexal V |
| |
| |
| The incorporation of viral membrane proteins or peptide antigens into liposomes has been shown to potentiate cell-mediated and humoral immune response and generate solid and durable immunity against the pathogen. Virosomes are reconstituted virus liposomes, constructed without the genetic information of the virus making them unable to replicate or cause infection [23-24]. Epaxal and Inflexal V are both vaccine products using the virosome-based antigen delivery system for commercial use (Table 1). For the production of Inflexal V, the influenza viruses, grown in hens’ eggs, are first inactivated with beta-propiolactone. The influenza surface antigens, Hemagglutinin (HA) and Neuraminidase (NA), are then purified and mixed with the phospholipid lecithin to form virosomes. Due to the virosomal technology, Hepatitis A Virus (HAV) vaccine Epaxal® and influenza vaccine Inflexal® V are highly efficacious by mimicking natural viral infection. The use of virosomes to deliver hepatitis A or influenza antigens stimulates a strong immune response of immuno competent cells. In contrast to other commercially available Hepatitis A Virus (HAV) vaccines, Epaxal is an aluminium-free vaccine based on formalin-inactivated hepatitis A (strain RG-SB) antigen incorporated virosomes. In clinical study by Usonis et al. [66], seroprotection rates were 100% in all infants and children at 1 and 12 months after primary vaccination with Epaxal. In contrast, the seroprotection rate after vaccination with aluminium containing vaccine Havrix was 67.7% in infants with pre-existing maternal anti-HAV antibodies, and a booster vaccination was required for complete seroprotection. Moreover, Epaxal was generally well tolerated by infants and children, with no serious systemic or local events reported after either primary or booster vaccination. |
| |
| For Inflexal V, most studies have shown interior efficacy or ineffectiveness on clinical parameters for these vaccines compared with the nonadjuvanted, split-virus or subunit seasonal vaccines [67]. Kanra et al. [68] compared the immunogenicity and safety of Inflexal V in children with a split influenza vaccine, Fluarix. Both vaccines were well tolerated and could induce effective immune responses in children. Interestingly, the virosome-adjuvanted influenza vaccine showed greater immunogenicity (88.8% seroconversion rates for H3N2) over the split influenza vaccine (77.5% seroconversion rates for H3N2) in unprimed children. In essence, virosomal techniques may not be able to give superior protective immunity in clinic but it has given humankind the time to prepare for a potential public health inflection. |
| |
| Liposomal formulations in ophthalmology: Visudyne |
| |
| Verteporfin is a hydrophobic chlorin-like photosensitizer, which has been shown to be a highly effective for photodynamic therapy in vivo. However, Verteporfin also has a tendency to undergo self aggregation in aqueous media, which can severely limit drug bioavailability to biological systems. It is important to introduce verteporfin into the bloodstream in its monomeric form and hence verteporfin was encapsulated in liposomes (Visudyne) for intravenous drug delivery [19-21]. The lipid layers of Visudyne are composed of unsaturated Egg Phosphatidyl Glycerol (EPG) and Dimyristoyl Phosphatidyl Choline (DMPC) in 3:5 molar ratio. Visudyne was only drug approved by the FDA for the photodynamic treatment of agerelated macular degeneration. Visudyne treatment prevents the growth of the destructive blood vessels without hurting the surrounding tissues. Phase I and II clinical studies were conducted for 609 patients with age-related macular degeneration [69-70]. After 12 month treatment, the group treated with Visudyne (6 mg/m2 body surface area) had statistically better visual acuity, contrast sensitivity, and fluorescein angiographic outcomes than did those who had placebo treatment (5% dextrose in water). At the month-12 examination, 246 (61%) of 402 eyes assigned to verteporfin compared with 96 (46%) of 207 eyes assigned to placebo had lost fewer than 15 letters of visual acuity from baseline. In subgroup analyses, the visual acuity benefit of verteporfin therapy was clearly demonstrated (67% vs 39%) when the area of choroidal neovascularization, caused by age-related macular degeneration, occupied 50% or more of the area of the entire lesion. However, Visudyne was readily destabilized in the presence of relatively low concentrations of plasma as results reported by Chowdhary et al. [19]. Therefore, the future investigation of liposomal formulations in ophthalmology is to stable liposome structures for expending the plasma circulation time following intravenous injection. |
| |
| Conclusions |
| |
| Since the first liposomal pharmaceutical product, Doxil, received FDA approval in 1995, liposomes are widely applied as drug carriers in clinic. Until now, several important types of liposomes, like temperature sensitive liposomes (ThermoDox) and cationic liposomes (EndoTAG1-1) have been extensively studied for clinic use. New liposomal formulations may extend plasma circulation time, vary drug distribution in body and hence reduce the possible side effects related to the drugs. However, these new generation liposomes have only comparable or poor therapeutic efficiency to conventional vesicles in clinical trials. Therefore, new liposomal pharmaceutical products should focus more on the types of delivered drugs (from small hydrophobic anti-cancer drugs to influenza surface antigens) and therapeutic applications (from anti-cancer chemotherapy to vaccination) than formulation design. |
| |
| Acknowledgment |
| |
| This work was supported by the National Science Council (Taiwan) (grant no: NSC 98-2320-B-415-001-MY3). |
| |
| |
| References |
| |
- Jesorka A, Orwar O (2008) Liposomes: technologies and analytical applications. Annu Rev Anal Chem (Palo Alto Calif) 1: 801-832.
- Makino K, Shibata A (2006) Chapter 2: Surface properties of liposomes depending on their composition. Advances in Planar Lipid Bilayers and Liposomes 4: 49-77
- Irache JM, Esparza I, Gamazo C, Agüeros M, Espuelas S (2011) Nanomedicine: novel approaches in human and veterinary therapeutics. Vet Parasitol 180: 47-71.
- Allen TM, Moase EH (1996) Therapeutic opportunities for targeted liposomal drug delivery. Adv Drug Deliv Rev 21: 117-133.
- Immordino ML, Dosio F, Cattel L (2006) Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine 1: 297-315.
- Official website from Astellas Pharma US, Inc.
- Meunier F, Prentice HG, Ringdén O (1991) Liposomal amphotericin B (AmBisome): safety data from a phase II/III clinical trial. J Antimicrob Chemother 28: B83-B91.
- Official website from Enzon Pharmaceuticals, Inc.
- Wasan KM, Lopez-Berestein G (1996) Characteristics of lipid-based formulations that influence their biological behavior in the plasma of patients. Clin Infect Dis 23: 1126-1138.
- Denning DW, Lee JY, Hostetler JS, Pappas P, Kauffman CA, et al. (1994) NIAID Mycoses Study Group multicenter trial of oral itraconazole therapy for invasive aspergillosis. Am J Med 97: 135-144.
- Official website from Three Rivers Pharmaceuticals, LLC.
- Official website from Gilead Sciences, Inc.
- Rivera E (2003) Liposomal anthracyclines in metastatic breast cancer: clinical update. Oncologist 8: 3-9.
- Tomkinson B, Bendele R, Giles FJ, Brown E, Gray A, et al. (2003) OSI-211, a novel liposomal topoisomerase I inhibitor, is active in SCID mouse models of human AML and ALL. Leuk Res 27: 1039-1050.
- Park JW (2002) Liposome-based drug delivery in breast cancer treatment. Breast Cancer Res 4: 95-99.
- Hoarau D, Delmas P, David S, Roux E, Leroux JC (2004) Novel long-circulating lipid nanocapsules. Pharm Res 21: 1783-1789.
- Hong RL (2004) Liposmal anti-cancer drug researches the myth of long circulation. J Chinese Oncol Soc 20: 10-21.
- Gardikis K, Tsimplouli C, Dimas K, Micha-Screttas M, Demetzos C (2010) New chimeric advanced Drug Delivery nano Systems (chi-aDDnSs) as doxorubicin carriers. Int J Pharm 402: 231-237.
- Chowdhary RK, Shariff I, Dolphin D (2003) Drug release characteristics of lipid based benzoporphyrin derivative. J Pharm Pharm Sci 6: 13-19.
- Fahr A, van Hoogevest P, May S, Bergstrand N, S Leigh ML (2005) Transfer of lipophilic drugs between liposomal membranes and biological interfaces: consequences for drug delivery. Eur J Pharm Sci 26: 251-265.
- Official website from NovartisOphthalmics, Inc.
- Patil SD, Burgess DJ (2005) Liposomes, Design and manufacturing. In: Burgess DJ (2005) Injectable Dispersed Systems, Formulation, Processing and Performance. Taylor & Francis.
- Stegmann T, Morselt HW, Booy FP, van Breemen JF, Scherphof G, et al. (1987) Functional reconstitution of influenza virus envelopes. EMBO J 6: 2651-2659.
- Glück R, Mischler R, Brantschen S, Just M, Althaus B, et al. (1992) Immunopotentiating reconstituted influenza virus virosome vaccine delivery system for immunization against hepatitis A. J Clin Invest 90: 2491-2495.
- D'Acremont V, Herzog C, Genton B (2006) Immunogenicity and safety of a virosomal hepatitis A vaccine (Epaxal) in the elderly. J Travel Med 13: 78-83.
- Herzog C, Hartmann K, Künzi V, Kürsteiner O, Mischler R, et al. (2009) Eleven years of Inflexal V-a virosomal adjuvanted influenza vaccine. Vaccine 27: 4381-4387.
- Zhang JA, Anyarambhatla G, Ma L, Ugwu S, Xuan T, et al. (2005) Development and characterization of a novel Cremophor EL free liposome-based paclitaxel (LEP-ETU) formulation. Eur J Pharm Biopharm 59: 177-187.
- Michaelis U, Haas H (2007) Targeting of Cationic Liposomes to Endothelial Tissue. In: Gregory Gregoriadis (2007) Liposome Technology: Interactions of Liposomes with the Biological Milieu Volume 3, Informa Healthcare.
- Eichhorn ME, Becker S, Strieth S, Werner A, Sauer B, et al. (2006) Paclitaxel encapsulated in cationic lipid complexes (MBT-0206) impairs functional tumor vascular properties as detected by dynamic contrast enhanced magnetic resonance imaging. Cancer Biol Ther 5: 89-96.
- Campbell RB, Ying B, Kuesters GM, Hemphill R (2009) Fighting cancer: from the bench to bedside using second generation cationic liposomal therapeutics. J Pharm Sci 98: 411-429.
- Li Z, Zhang Y, Wurtz W, Lee JK, Malinin VS, et al. (2008) Characterization of nebulized liposomal amikacin (Arikace) as a function of droplet size. J Aerosol Med Pulm Drug Deliv 21: 245-254.
- Mossalam M, Dixon AS, Lim CS (2010) Controlling subcellular delivery to optimize therapeutic effect. Ther Deliv 1: 169-193.
- Rodriguez MA, Pytlik R, Kozak T, Chhanabhai M, Gascoyne R, et al. (2009) Vincristine sulfate liposomes injection (Marqibo) in heavily pretreated patients with refractory aggressive non-Hodgkin lymphoma: report of the pivotal phase 2 study. Cancer 115: 3475-3482.
- Krishna R, Webb MS, St Onge G, Mayer LD (2001) Liposomal and nonliposomal drug pharmacokinetics after administration of liposome-encapsulated vincristine and their contribution to drug tissue distribution properties. J Pharmacol Exp Ther 298: 1206-1212.
- Yarmolenko PS, Zhao Y, Landon C, Spasojevic I, Yuan F, et al. (2010) Comparative effects of thermosensitive doxorubicin-containing liposomes and hyperthermia in human and murine tumours. Int J Hyperthermia 26: 485-498.
- Dromi S, Frenkel V, Luk A, Traughber B, Angstadt M, et al (2007) Pulsed-high intensity focused ultrasound and low temperature-sensitive liposomes for enhanced targeted drug delivery and antitumor effect. Clin Cancer Res 13: 2722-2727.
- Zahid S, Brownell I (2008) Repairing DNA damage in xeroderma pigmentosum: T4N5 lotion and gene therapy. J Drugs Dermatol 7: 405-408.
- Tari AM, Gutiérrez-Puente Y, Monaco G, Stephens C, Sun T, et al. (2007) Liposome-incorporated Grb2 antisense oligodeoxynucleotide increases the survival of mice bearing bcr-abl-positive leukemia xenografts. Int J Oncol 31: 1243-1250.
- Pal A, Khan S, Wang YF, Kamath N, Sarkar AK, et al. (2005) Preclinical safety, pharmacokinetics and antitumor efficacy profile of liposome-entrapped SN-38 formulation. Anticancer Res 25: 331-341.
- Dragovich T, Mendelson D, Kurtin S, Richardson K, Von Hoff D, et al. (2006) A Phase 2 trial of the liposomal DACH platinum L-NDDP in patients with therapy-refractory advanced colorectal cancer. Cancer Chemother Pharmacol 58: 759-764.
- Website of ClinicalTrials.gov
- Powell E, Chow LQ (2008) BLP-25 liposomal vaccine: a promising potential therapy in non-small-cell lung cancer. Expert Rev Respir Med 2: 37-45.
- Stathopoulos GP, Boulikas T (2012) Lipoplatin formulation review article. J Drug Deliv 2012: 581363.
- Duffaud F, Borner M, Chollet P, Vermorken JB, Bloch J, et al. (2004) Phase II study of OSI-211 (liposomal lurtotecan) in patients with metastatic or loco-regional recurrent squamous cell carcinoma of the head and neck. An EORTC New Drug Development Group study. Eur J Cancer 40: 2748-2752.
- Tomkinson B, Bendele R, Giles FJ, Brown E, Gray A, et al. (2003) OSI-211, a novel liposomal topoisomerase I inhibitor, is active in SCID mouse models of human AML and ALL. Leuk Res 27: 1039-1050.
- Semple SC, Leone R, Wang J, Leng EC, Klimuk SK, et al. (2005) Optimization and characterization of a sphingomyelin/cholesterol liposome formulation of vinorelbine with promising antitumor activity. J Pharm Sci 94: 1024-1038.
- Shaji J, Patole V (2008) Protein and Peptide drug delivery: oral approaches. Indian J Pharm Sci 70: 269-277.
- Meure LA, Foster NR, Dehghani F (2008) Conventional and dense gas techniques for the production of liposomes: a review. AAPS PharmSciTech 9: 798-809.
- Lesoin L, Boutin O, Crampon C, Badens E (2011) CO2/water/surfactant ternary systems and liposome formation using supercritical CO2: a review. Colloids Surf A Physicochem Eng Asp 377: 1-14.
- Castor TP (2005) Phospholipid nanosomes. Curr Drug Deliv 2: 329-340.
- Zagrobelny JA, Bright FV (1992) In situ studies of protein conformation in supercritical fluids: trypsin in carbon dioxide. Biotechnol Prog 8: 421-423.
- Uhumwangho MU, Okor RS (2005) Current trends in the production and biomedical applications of liposomes: a review. Pak J Pharm Sci 4: 9-21.
- Chen C, Han D, Cai C, Tang X (2010) An overview of liposome lyophilization and its future potential. J Control Release 142: 299-311.
- EL-Neser OH, Yahiya SA, EL-Gazayerly ON (2010) Effect of formulation design and freeze-drying on propertiesof fluconazole multilamellar liposomes. Saudi Pharmaceutical Journal 18: 217-224.
- Clemons KV, Stevens DA (1998) Comparison of fungizone, Amphotec, AmBisome, and Abelcet for treatment of systemic murine cryptococcosis. Antimicrob Agents Chemother 42: 899-902.
- Wu SY, Putral LN, Liang M, Chang HI, Davies NM, et al. (2009) development of a novel method for formulating stable siRNA-loaded lipid particles for in vivo use. Pharm Res 26: 512-522.
- Lammers T, Hennink WE, Storm G (2008) Tumour-targeted nanomedicines: principles and practice. Br J Cancer 99: 392-397.
- Mills JK, Needham D (2005) Lysolipid incorporation in dipalmitoylphosphatidylcholine bilayer membranes enhances the ion permeability and drug release rates at the membrane phase transition. Biochim Biophys Acta 1716: 77-96.
- Banno B, Ickenstein LM, Chiu GN, Bally MB, Thewalt J, et al. (2010) The functional roles of poly(ethylene glycol)-lipid and lysolipid in the drug retention and release from lysolipid-containing thermosensitive liposomes in vitro and in vivo. J Pharm Sci 99: 2295-2308.
- Gill PS, Wernz J, Scadden DT, Cohen P, Mukwaya GM, et al. (1996) Randomized phase III trial of liposomal daunorubicin versus doxorubicin, bleomycin, and vincristine in AIDS-related Kaposi's sarcoma. J Clin Oncol 14: 2353-2364.
- Ratna Mehta R, Thomas G. Burke TG (2004) Membrane Biophysical Parameters Influencing Anthracycline Action. In: Priebe W (2004) Anthracycline Antibiotics. ACS Publication.
- Fetterly GJ, Straubinger RM (2003) Pharmacokinetics of paclitaxel-containing liposomes in rats. AAPS PharmSci 5: E32.
- Fetterly GJ, Grasela TH, Sherman JW, Dul JL, Grahn A, et al. (2008) Pharmacokinetic/pharmacodynamic modeling and simulation of neutropenia during phase I development of liposome-entrapped paclitaxel. Clin Cancer Res 14: 5856-5863.
- Winer EP, Berry DA, Woolf S, Duggan D, Kornblith A, et al. (2004) Failure of higher-dose paclitaxel to improve outcome in patients with metastatic breast cancer: cancer and leukemia group B trial 9342. J Clin Oncol 22: 2061-2068.
- Bayraktar S, Bayraktar UD, Rocha-Lima CM (2010) Recent developments in palliative chemotherapy for locally advanced and metastatic pancreas cancer. World J Gastroenterol 16: 673-682.
- Usonis V, Bakasénas V, Valentelis R, Katiliene G, Vidzeniene D, et al. (2003) Antibody titres after primary and booster vaccination of infants and young children with a virosomal hepatitis A vaccine (Epaxal). Vaccine 21: 4588-4592.
- Nicholson KG, Thompson CI, Klap JM, Wood JM, Batham S, et al. (2009) Safety and immunogenicity of whole-virus, alum-adjuvanted whole-virus, virosomal, and whole-virus intradermal influenza A/H9N2 vaccine formulations. Vaccine 28: 171-178.
- Kanra G, Marchisio P, Feiterna-Sperling C, Gaedicke G, Lazar H, et al. (2004) Comparison of immunogenicity and tolerability of a virosome-adjuvanted and a split influenza vaccine in children. Pediatr Infect Dis J 23: 300-306.
- Bressler NM, Bressler SB (2000) Photodynamic therapy with verteporfin (Visudyne): impact on ophthalmology and visual sciences. Invest Ophthalmol Vis Sci41: 624-628.
- (1999) Photodynamic therapy of subfoveal choroidal neovascularization in age-related macular degeneration with verteporfin: one-year results of 2 randomized clinical trials-TAP report. Treatment of age-related macular degeneration with photodynamic therapy (TAP) study group. Arch Ophthalmol 117: 1329-1345.
|
| |
| |