|
|
| Rosaiah Kanaparthy1* and Aruna Kanaparthy2 |
| 1Department of Periodontics, Peoples Dental Academy, Bhopal, Madhya Pradesh, India |
| 2Conservative Dentistry, Peoples Dental Academy, Bhopal, Madhya Pradesh, India |
| *Corresponding authors: |
Rosaiah Kanaparthy
Department of Periodontics, Peoples Dental Academy
HIG-3, PDA Staff Quarters, Peoples Campus
Bhanpur, Bhopal, Madhya Pradesh, India
Tel: 91 9893050554
E-mail: medha98@gmail. com |
|
| |
| Received June 19, 2012; Published July 26, 2012 |
| |
| Citation: Kanaparthy R, Kanaparthy A (2012) Craniofacial Dysostosis-The Dental Perspective: A Case Report. 1: 196. doi:10.4172/scientificreports.196 |
| |
| Copyright: © 2012 Kanaparthy R, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| The Crouzon syndrome is a rare clinical condition that affects the craniofacial skeleton development. Although it is uncommon, it has a transmission risk of 50% when one of the parents is a carrier of defective or mutant gene. The dominant transmission range is of 100% and the gene penetrance and its phenotypic expression has been observed to be highly variable. It accounts for about 4.8% of all the cases of craniosynostosis, and it is the most common syndrome presenting craniosynostoses. The Crouzon syndrome’s early diagnosis is critical to avoid cranial hypertension, as well as visual disturbances and blindness. Therefore, it is important to pay close attention to the patients that have some Crouzon syndrome carrier relative precedent or that have a certain level of exophthalmia. Children who have Crouzon syndrome have a range of problems of variable severity, from mild facial symptoms causing a primarily cosmetic concern, to severe symptoms affecting breathing, feeding, vision and brain development. Functional, esthetic and psychological reasons make it important to start treatment early in life. The dental profession should have sufficient knowledge of syndromes associated with dysmorphic facies to detect patients who are unaware of their condition. This case report emphasizes the role of a dentist in diagnosing such conditions and co-ordinating a multi-disciplinary team for corrective measures. The team is made complete by the geneticist, psychologist, speech and language therapist, respiratory care specialist, and specialist nursing staff. |
| |
| Keywords |
| |
| Craniosynostoses; Crouzon; Craniofacial dysostosis; Exophthalmia |
| |
| Introduction |
| |
| This syndrome was originally described in 1912 by a French neurologist. He described four essential characteristics: exorbitism, retromaxillism, inframaxillism and parodoxic retrognathia. The incidence of this syndrome appears to be approximately one in 25,000 in the general population. It is inherited as an autosomal dominant pattern with variable expression. |
| |
| The mutation in the genes that codify type 2 fibroblast growth factor receptor (FGFR2), is responsible for the deformities observed. However, 50% of the Crouzon syndrome new cases are not inherited but result from new spontaneous mutations [1-6]. Crouzon syndrome is characterized by craniosynostosis, maxillary hypoplasia, shallow orbits with proptosis and bifid uvula. There are often intracranial abnormalities such as anomalous venous drainage and hydrocephalus. A variety of other manifestations of Crouzon syndrome exist, including calcification of the stylohyoid ligament in 50% of patients over 4 years old, cervical spine abnormalities in up to 40% of patients, elbow malformations (18%), minor hand deformities (10%), visceral anomalies (7%), various musculoskeletal deformities (7%), and skin lesions . Stylohyoid ligament calcification is also reported in 38%-88% of cases of Apert syndrome . Cervical spine fusion anomalies affecting C2 to C5 are the most common vertebral deformities in Crouzon syndrome. Limb anomalies in Crouzon syndrome are nonspecific. Acanthosis nigrans (hyperpigmented, hyperkeratotic lesions located on the neck and near joint flexures) has also been reported in Crouzon syndrome [7,8]. |
| |
| Treatment of craniofacial syndromes consists of a team approach in which each organ system is addressed independently and problems are prioritized on the basis of relative urgency. |
| |
| Presentation of Case |
| |
| A 24 year old female patient reported to the dental clinic with the chief complaint of pain in 36 since the past 2 days and also expressed concern about her difficulty in mastication and lack of dental and facial harmony. The patient’s sister also had similar problems but was not available for examination. Medical history was insignificant and the patient appeared to have normal mental faculties. |
| |
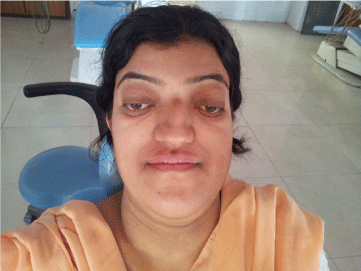
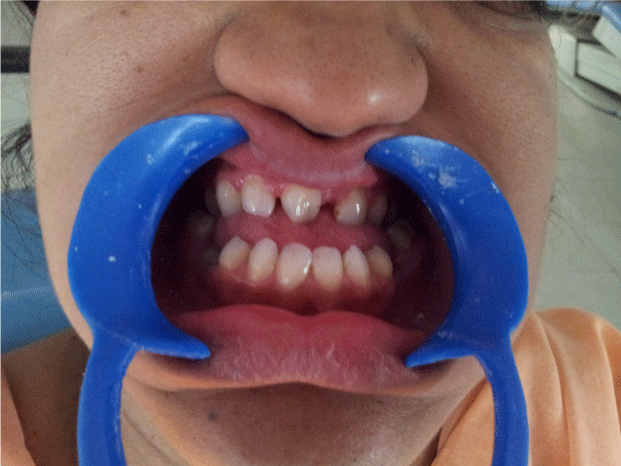
| On clinical examination, the patient presented with the typical manifestations of craniofacial dysostosis. Brachycephalic head form, prominent forehead, concave profile, decreased molar prominence, ocular proptosis, hypoplastic midface, exophthalmos hypertelorism , beak like nose (psittichornia), low set ears excessive lower facial height, maxillary retrognathism and a short upper lip (Figure 1-3). Intraoral examination revealed-Class III dental relationship, narrow, v-shaped maxilla, high-arched palate, bilateral posterior crossbite, spacing between upper anterior teeth malocclusion, oligodontia with missing 21, 32, 42, 35, 45, 47, submerged, retained deciduous 85, tender to touch 36 with deep caries. Patient gave history of recent extraction of retained 75. Periodontal status was normal. |
| |
|
|
Figure 1: Front view of patient showing Brachycephalic head form, prominent forehead, decreased malar prominence, ocular proptosis, hypoplastic midface , hypertelorism, beak like nose (psittichornia). |
|
| |
|
|
Figure 2: Lateral view shows concave profile, exophthalmos, low set ears, excessive lower facial height, maxillary retrognathism and short upper lip. |
|
| |
|
|
Figure 3: Intra oral view shows missing 21, 32, 42, 35, 45 and retained 85. |
|
| |
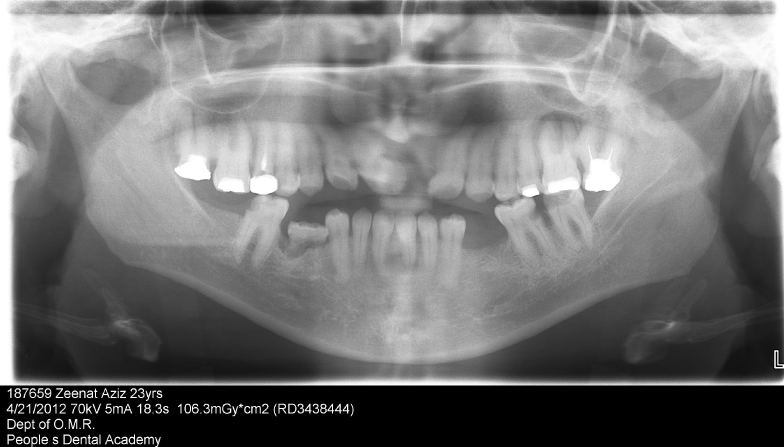
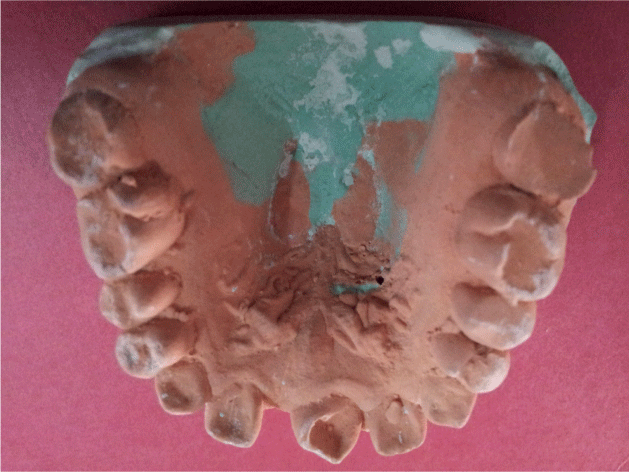
| The set of records obtained were study casts, posteroanterior skull, wrist and panoramic radiographs, and photographs. The skull radiograph presented with a copper-beaten appearance, wrist radiographs were normal. The panoramic radiograph showed retained deciduous submerged tooth 85, tilted teeth, edentulous space in the third quadrant, missing 21, 32, 35, 42, 45, 47 along with periapical lesions in relation to 26 and 31 (Figure 4-6). |
| |
|
|
Figure 4: Dental Cast shows narrow, v-shaped maxilla, high-arched palate, upper anterior spacing , missing 21. |
|
| |
|
|
Figure 5: OPG shows missing 21, 32, 42, 35, 45, 47, retained deciduous 85, spacing between upper anteriors, edentulous area in III quadrant, periapical radiolucencies in relation to 26, 31and deep caries in relation to 36. |
|
| |
|
|
Figure 6: The skull radiograph showed a copper-beaten appearance. |
|
| |
| Treatment planning |
| |
| The diagnosis for 36 was irreversible pulpitis and endodontic treatment was initiated. The patient was counseled in regard to the craniofacial condition she was suffering from and treatment possibilities explained. She was referred to a plastic surgeon, an oral surgeon an ophthalmologist an orthodontist and a prosthodontist working independently for further evaluation and action. |
| |
| Discussion and Conclusion |
| |
| Crouzon syndrome is inherited as a highly variable autosomal dominant condition, and approximately half the cases are familial. Approximately 30 different mutations of fibroblast growth factor receptor II have been identified in Crouzon syndrome to date. The Crouzon phenotype is highly variable and ranges from ocular proptosis and midface hypoplasia with no craniosynostosis to a cloverleaf skull deformity. Unlike most other craniofacial syndromes caused by fibroblast growth factor receptor mutations, the limbs are typically unaffected [9].The fibroblast growth factors are intrinsically related to the extracellular matrix formation. When the extracellular matrix presents FGFR2’s mutation, it begins to secrete cytokines both in autocrinous and paracrinous manner and these may modify the very matrix. It is allegeable that such changes are in the osteogenic process change genesis, which explains the pathologic variations found. In this disease, the premature closure of cranial sutures and midfacial sutures give it a branchiocephalic configuration [10,11-14]. There is an underdeveloped mid face, and receded cheekbones. The patients appear exopthalmic due to the shallow orbits. They exhibit an Angle Class III malocclusion due to the midface deficiency, while the mandibular growth potential is normal. Because of the underdeveloped midface and high arched palate, nasal airway obstruction may occur. |
| |
| The obstruction of the upper respiratory passages develops, following the septal diversion, abnormalities to the center of the nose and epipharynx narrowing. It can lead to acute respiratory anxiety, dyspnea of the type polypnea and even sleep apnea, mainly when connected to upper maxillary hypoplasia [14]. Other features commonly encountered are visual disturbances due to eye muscle imbalance and frequent hearing loss due to recurrent ear infections. The conductive hearing loss is common due to the medium ear deformities [14]. |
| |
| There are several ocular abnormalities and the most common already reported for such disease are: shallow orbits, bilateral ocular proptosis, hypertelorism, divergent strabismus, optical atrophy, conjunctivitis or exposure keratoconjunctivitis and a non-explained loss of visual accuracy . There rarely may occur nystagmus, coloboma of the iris, anisocoria, microcornea or megalocornea, cataract, blue sclerae, glaucoma and globe luxation. Blindness following the optical atrophy by the intracranial hypertension may also occur [14]. Spectaclesplasty enblocrotation advancement of the periorbital bony skeleton can be safely performed before skeletal maturity which produces a more normal anatomic position of the periorbital soft tissues facilitating both function and aesthetics [9]. Mental capacity is usually normal. |
| |
| An early craniosynostosis, evidenced by the existence of intracranial hypertension, is present in 60% of the cases and furnishes a reserved visual prognostic. The patients have hyperemia, bilateral ocular irritation and sensation of long-term nuisance for being constantly rubbed [14]. Diagnosis of Crouzon syndrome usually can occur at birth by assessing the signs and symptoms of the baby. Further analysis, including radiographs, magnetic resonance imaging (MRI) scans, genetic testing, X-rays and CT scans can be used to confirm the diagnosis of Crouzon syndrome [7]. Treatment of Crouzon syndrome is complex, since there are many aspects of the syndrome to manage. |
| |
| Crouzon syndrome almost always requires surgery to enable the skull to expand properly and to align the jaws, along with numerous other surgeries to repair face and ear defects. The entire middle portion of the face may be surgically extended forward by a number of surgical techniques. Once treated for the cranial vault symptoms, Crouzon patients generally go on to live a normal lifespan [14,15]. |
| |
| Conflict of Interest |
| |
| No potential conflict of interest is relevant to this article, all authors declare that no financial and personal relationships with other people or organizations could inappropriately influence (bias) their work. Examples of potential conflicts of interest include employment, consultancies, stock ownership, honoraria, paid expert testimony, patent applications/registrations, and grants or other funding. |
| |
| |
| References |
| |
- Bowling EL, Burstein FD (2006) Crouzon syndrome. J Am Optom Assoc 77: 217-222.
- Eswarakumar VP, Horowitz MC, Locklin R, Morriss-Kay GM, Lonai P (2004) A gain-of- function mutation of Fgfr2c demonstrates the roles of this receptor variant in osteogenesis. Proc Natl Acad Sci U S A 101: 12555-12560.
- Hoefkens MF, Vermeij-Keers C, Vaandrager JM (2004) Crouzon Syndrome: Phenotypic Signs and Symptoms of the Postnatally expressed Subtype. J Craniofac Surg 15: 233-240.
- Rice DP (2008) Clinical features of syndromic craniosynostosis. Front Oral Biol 12: 91-106.
- Cohen MM Jr, Kreiborg S (1992) Birth prevalence studies of the Crouzon syndrome: comparison of direct and indirect methods. Clin Genet 41: 12-15.
- Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, et al. (1994) Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet 8: 98-103.
- Lowe LH, Booth TN, Joglar JM, Rollins NK (2000) Midface anomalies in children.Radiographics 20: 907-922.
- Proudman TW, Moore MH, Abbott AH, David DJ (1994) Noncraniofacial manifestations of Crouzon’s disease. J Craniofac Surg 5: 218-222.
- Kreiborg S (1981) Craniofacial growth in plagiocephaly and Crouzon syndrome. Scand J Plast Reconstr Surg 15: 187-197.
- Carinci F, Pezzetti F, Locci P, Becchetti E, Carls F, et al. (2005) Apert and Crouzon Syndromes: Clinical Findings, Genes and Extracellular Matrix. J Craniofac Surg 3: 361-368.
- Ousterhout DK, Zlotow IM (1990) Aesthetic improvement of the forehead utilizing methylmethacrylate onlay implants. Aesthetic Plast Surg 14: 281-285.
- Crouzon Craniofacial Center 1: 418-3223.
- Singer SL, Walpole I, Brogan WF, Goldblatt J (1997) Dentofacial features of a family with Crouzon syndrome. Case reports. Aust Dent J 42: 11-17.
- Silva DLD, Neto P, Xavier F, Carneiro SG, Palheta ACP, et al. (2008) Cruouzon’s syndrome: Literature review. International Arch Otorhinolaryngology 12: 436-441.
- Craniofacial dysostosis, Wikepedia.
|
| |
| |