|
|
| Krati Shah* |
| Clinical Genetics Unit, Christian Medical College, Vellore, India |
| *Corresponding authors: |
Krati Shah
Post-Doctoral Fellow, Clinical Genetics Unit
Christian Medical College, Vellore, India
Tel: 09751692983
E-mail: kratishah@yahoo.co.in |
|
| |
| Received August 21, 2012; Published August 25, 2012 |
| |
| Citation: Shah K (2012) Yet Another Case of Typical Hallerman Streiff Syndrome Without Mutations in the GJA1 Gene. 1: 245. doi:10.4172/scientificreports.245 |
| |
| Copyright: © 2012 Shah K. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Introduction |
| |
| Hallermann-Streiff syndrome (HSS) was first time described by Hallerman in 1948 and Streiff in 1950 who distinguished it from progeria and mandibulofacial dysostosis. Disease causing pathogenic mutation in GJA1 gene has been identified in a patient with clinical features that overlap Hallerman-Streiff syndrome (HSS; MIM# 234100) with Oculodentodigital dysplasia (ODDD; MIM# 164200) [1] However none of the cases with typical HSS are found to contain mutations in the GJA1 gene. We describe an Indian patient with typical HSS first time without identifiable GJA1 mutations. |
| |
| Clinical Brief |
| |
| Seven year old girl of Asian- Indian origin presented to us with complaints of short stature and sparse scalp hair. She is the first child of non-consanguineous marriage. Mother’s and father’s age were 23 and 25 years respectively at the time of birth. During antenatal period mother had gestational diabetes mellitus controlled with dietary advice and medications. Proposita was born by caesarian section with birth weight of 2.5 kg (5th centile). She was operated for bilateral eye cataract at one month of age. Her vision is 6/6 both eyes (Snellen visual acuity chart) with appropriate refractive correction. Her parents complained of snoring from neonatal age. She developed obstructive sleep apnea syndrome and was prescribed positive pressure ventilation at nights. She studies in class appropriate for her age and is good in studies. She has a normal younger brother of two years age. |
| |
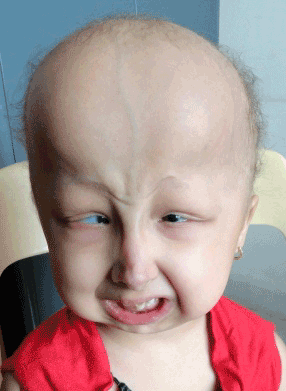
| The proposita had proportionate short stature with height 87 cm (<5 centile) and weight 10 kgs (<5 centile). She has the following dysmorphic features (Figure 1): brachycephalic with frontal bossing (head circumference 48 cm), prominent scalp veins, sparse thin and curly scalp hair, sparse eyebrows with no eye lashes or body hair, microphthalmia with blue sclera, down slanting palpebral fissures with telecanthus, beaked nose with hypoplastic alae nasi, abnormal dentition leading to dental caries and micrognathia. The patient fulfilled the criteria of HSS and GJA1 gene analysis was carried out. |
| |
|
|
Figure 1: Facial profile of proband. |
|
| |
| Methods |
| |
| Informed written consent was taken and blood was collected for molecular analysis. DNA was extracted with Qiagen midi kit following manufacturer instructions. The coding regions of GJA1 gene were amplified by PCR using primers described by Paznekas et al. [2]. GJA1 gene comprises of two exons. Forward primer 5'-GATCTTTTCTTCGTTGGC-3' (within intron 1) and reverse primer 5'-CTCTTTCCCTTAACCCG-3' yielded 925 bp product. Another set of primers 5'-TTCCTCTCTCGCCCCAC-3' and 5'-GGCCTAGAAAGCTTACCTT-3' (extend into the 3' UTR) yielded 1,079-bp product. PCR conditions were standardized for both sets of primers. PCR products were sequenced in the forward and reverse directions on an ABI prism 3130 automated sequencer. Alw44I restriction enzyme analysis was done for GJA1 c.227G>A mutation. |
| |
| Results |
| |
| Restriction enzyme digestion failed to show the mutation reported by Pizzuti et al. [1]. Sequence analysis of the GJA1 gene showed normal sequence. |
| |
| Discussion |
| |
| HSS (#234100) is characterized by craniofacial deformity with eye abnormality, proportionate short stature and hypotrichosis. Our patient had all seven diagnostic features and was thus “full-blown” phenotype. Snaepen et al. [3] describes three cases of HSS and mentions it as a classification overlap with ODDD. |
| |
| Homozygous GJA1 gene mutation has been reported by Pizzuti et al. [1] in one of the HSS/ODDD overlap cases reported by Snappen. This case was rather a misdiagnosis of HSS in a case of ODDD [4]. |
| |
| GJA1, a gene for Cx43 is described as causative factor for ODDD [2]. GJA1 codes for connexin-43, a transmembrane protein that facilitate cell-to-cell adhesion and provide pathways for direct intercellular communication. They help in cell adhesion and migration. They are expressed in various tissues such as the heart, liver, lymphoctes, limbs etc. and play a key role in tissue homeostasis and regulation of growth, development, and differentiation. Defect in connexin availability at different period of embryogenesis might lead to developmental anomalies of organs pertaining to the stage of development [5]. |
| |
| Including this study GJA1 screening has been performed in eight well-characterized patients with HSS. Paznekas et al. [2] did molecular analysis of six patients with typical HSS and no changes in the sequences of the GJA1 gene were found. None of the patients including ours diagnosed with HSS have been found to have identifiable mutation. Though a possibility of a variation in the non-coding region of the gene that affects the expression of the gene remains, it is more likely that HSS does not come under GJA1 spectrum of disease phenotype and a different gene is responsible for this disease. Typical cases of HSS should have GJA1 gene analysis to further support this. Other candidate genes should be searched for by newer sequencing technology. |
| |
| Acknowledgement |
| |
| We thank the family for giving consent for photography and participating in the study. |
| |
| |
| References |
| |
- Pizzuti A, Flex E, Mingarelli R, Salpietro C, Zelante L, et al. (2004) A homozygous GJA1 gene mutation causes a Hallermann-Streiff/ODDD spectrum phenotype. Hum Mutat 23: 286.
- Paznekas WA, Karczeski B, Vermeer S, Lowry RB, Delatycki M, et al. (2009) GJA1 mutations, variants, and connexin 43 dysfunction as it relates to the oculodentodigital dysplasia phenotype. Hum Mutat 30: 724-733.
- Spaepen A, Schrander-Stumpel C, Fryns JP, de Die-Smulders C, Borghgraef M, et al. (1991) Hallermann-Streiff syndrome: clinical and psychological findings in children. Nosologic overlap with oculodentodigital dysplasia? Am J Med Genet. 41: 517-520.
- Gorlin RJ, Cohen MM, Hennekam RCM (2001) Syndromes of the Head and Neck Chapter 10 (4th edn).
- Lo CW (1996) The role of gap junction membrane channels in development. J Bioenerg Biomembr 28: 379-385.
|
| |
| |