| Research Article |
Open Access |
|
| Tapas Kumar Laha1*, Gitanjali Mishra2 and Subrata Sen1 |
| 1Acharya Prafulla Chandra Ray Memorial Cancer Chemotherapeutic Research Unit, College of Pharmaceutical Sciences, Mohuda, Ganjam, Berhampur-760002, Orissa, India |
| 2Berhampur University, Bhanja Bihar, Berhampur(Gm) -760007, Orissa, India |
| *Corresponding author: |
Tapas Kumar Laha
Acharya Prafulla Chandra Ray Memorial Cancer Chemotherapeutic Research Unit, College of Pharmaceutical Sciences, Mohuda
Ganjam, Berhampur-760002, Orissa, India
E- mail: tapaslaha80@rediffmail.com |
|
| |
| Received August 10, 2012; Published September 08, 2012 |
| |
| Citation: Laha TK, Mishra G, Sen S (2012) A Validated Stability Indicating Reversed Phase High Performance Liquid Chromatographic Method of Duloxetine and Characterization of its Degradation Products through Retro-Synthesis. 1:308. |
| |
| Copyright: © 2012 Laha TK, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| The present paper deals with the development of stability indicating reversed phase high-performance liquid chromatographic (RP-HPLC) method for duloxetine, an antidepressant drug in presence of its degradation products formed during forced decomposition studies. Forced degradation studies were performed on the bulk drug by using acid (0.1 N hydrochloric acid), base (0.1 N sodium hydroxide), water (neutral hydrolysis), 3% v/v hydrogen peroxide (oxidation), dry heat (60°C) and UV light (254 nm). Degradation was observed for duloxetine in all cases except dry heat and UV light and the formed degradation products were found to be 1-naphthol (degradation product-1), N-methyl-3-hydroxy-3-(2-thienyl) propylamine (degradation product-2) and duloxetine succinamide (degradation product-3). Successful separation of the drug from the degradation products formed under different stress conditions was achieved on a μ-Bondapak C18 column (250mm ×4.6mm, 5μm particle size) using methanol- phosphate buffer (pH 7.8; 50 mM) (7:3, v/v) as the mobile phase at a flow rate of 1.5 ml/min. The detection wavelength was 221nm. The developed method was completely validated and proved to be robust. As the method could effectively separate the drug from its degradation products, it can be employed for analysis of the samples of stability study. |
| |
| Keywords |
| |
| HPLC; Duloxetine, stress testing, stability-indicating, degradation products. |
| |
| Introduction |
| |
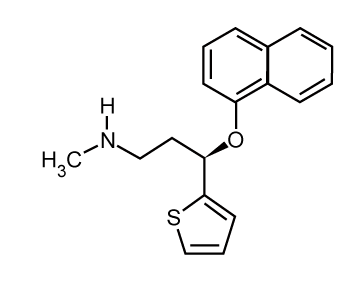
| Duloxetine (N-Methyl-3-naphthlen-yloxy-3-thiophen-2-yl-propan- 1-amine) [1] a selective serotonin and norepinephrine reuptake inhibitor (SSNRI) is used for the treatment of major depressive disorder and anxiety [2-4]. It is used for the treatment of neuropathic pain associated with peripheral neuropathy especially diabetic polyneuropathy for which it is first-line, and as an add-on treatment in stress urinary incontinence instead of surgery [5-6] also indicated for the management of fibromyalgia [7-8]. It restores the balance of neurotransmitters in the brain like serotonin and norepinehrine [9]. Moreover it is also being used in the treatment of peripheral neuropathy caused by certain anti cancer drugs [10]. |
| |
| A literature survey indicated few methods for the determination of duloxetine and its key intermediate, desmethyl-duloxetine, in human serum by HPLC [11-12]. Reports were found regarding the characterization of phenolic impurities in duloxetine samples by MS, NMR spectrometry and X-ray analysis [13] and of impurities formed by interaction of duloxetine with various enteric polymers [14] A simple UV spectrophotometric method for the estimation of duloxetine in a formulation was reported [15]. An HPLC method to separate duloxetine and structurally related impurities using a combination of computer-based solvent strength optimization and solvent selectivity mixture design [16]. An HPTLC method for estimation of duloxetine in bulk and in tablet dosage form [17]. A capillary electrophoresis with laser-induced fluorescence detection method also reported for estimation of duloxetine in human plasma [18]. During our literature survey, very few articles related to the stability-indicating HPLC determination of duloxetine were found [19-24] but no article related to the formation of N-methyl-3-hydroxy-3-(2-thienyl) propylamine, a potential degradation product of duloxetine was reported. Therefore the scientific novelty of the present work is to develop a simple, rapid, sensitive and less expensive method which can easily determine N-methyl-3-hydroxy-3-(2-thienyl) propylamine along with other degradation products reported. The stability assessment of any promising drug candidate plays a vital role in its preformulation study. Many environmental conditions such as heat, light, moisture as well as the inherent chemical susceptibility of a substance to hydrolysis or oxidation can play an important role in pharmaceutical stability. So, this study also helps to define storage and handling conditions. The exposition of the drug substance to extreme external conditions helps to reveal and identify the likely degradation products which will open a new scope of research on toxicity study. The findings of toxicity study will help in scrupulous determination of expiry, adverse effects etc. |
| |
| Experimental |
| |
| Apparatus |
| |
| Experiments were performed using a Waters (India) 510 HPLC system with Waters 486 tunable absorbance detector. The samples were injected manually using a 20 μl sample loop. The Millennium 32 software was used for quantification and data processing. |
| |
| Chemicals and reagents |
| |
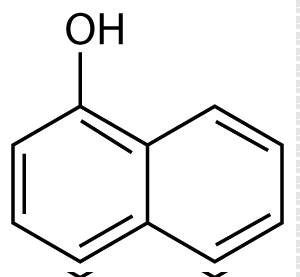
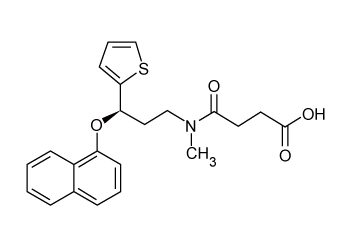
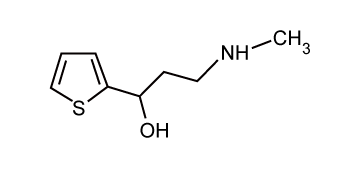
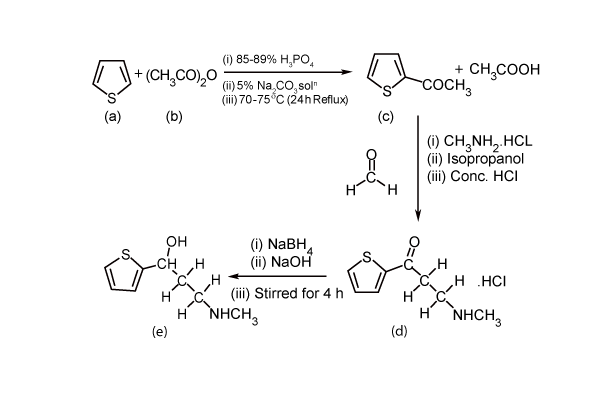
| Pure duloxetine (Figure 1) was provided by Wockhardt Limited, Mumbai, India and its degradation products namely 1-naphthol (Figure 2) was procured from Mayur Dye-Chem Intermediates Ltd., Gujarat, India, duloxetine succinamide (Figure 3) was purchased from Pro Lab Marketing Pvt. Ltd., New Delhi, India and N-methyl-3- hydroxy-3-(2-thienyl) propylamine (Figure 4) was synthesized through retro-synthetic pathway (Figure 5) in our own laboratory. Methanol and water of HPLC grade and were purchased from Merck (India) Ltd., Mumbai, India. All other chemicals and reagents used were of analytical grade and were purchased from Merck (India) Ltd., Mumbai, India. Two commercial formulations of duloxetine [DULOJOY (Torrent India Limited) and DUREEP (Alkem Laboratories Ltd.)] were used in this study. |
| |
|
|
Figure 1: N-Methyl-3-naphthlen-yloxy-3-thiophen-2-yl-propan-1-amine (Ranolazine dihydrochloride) |
|
| |
|
|
| |
|
|
Figure 3: N-methyl-3-hydroxy-3-(2-thienyl) propylamine. |
|
| |
|
|
Figure 4: Duloxetine succinamide. |
|
| |
|
|
Figure 5: Retro-synthetic pathway of N-methyl-3-hydroxy-3-(2-thienyl) propylamine. |
|
| |
| Retro-synthesis of N-methyl-3-hydroxy-3-(2-thienyl) propylamine |
| |
| Initially 2-Acetylthiophene was prepared by the acetylation of thiophene with acetic anhydride in the presence of orthophosphoric acid. Placed 13ml of thiophene and 9 ml of acetic anhydride in a 250ml three necked flask, fitted with a thermometer, mechanical stirrer and reflux condenser. Heated the stirred solution to 70-75 °C, removed the source of heat, added 2.5 ml of 87 percent orthophosphoric acid. An exothermic reaction occurred after 2-3 minutes and temperature raised up to 90 °C; Immersed the flask in a bath of cold water to control the reaction. When the boiling subsides (ca. 5 minutes) refluxed the mixture for 2 hrs at 175-190 °C, added 30 ml of water, stirred for 5 minutes, transferred the cold reaction mixture to a separatory funnel, removed the water layer, washed with two 15 ml portions of 5 percent sodium carbonate solution and dried over anhydrous magnesium sulphate. Distilled the orange-red liquid through a short fractionating column at atmospheric pressure and recovered 6.5 gram of unchanged thiophene at 83-84°C. Distilled the residue under reduced pressure and collect the 2-acetylthiophene at 89-90 °C/10mm; this solidifies on cooling in ice (yield 8.18gm) with this 6.66gm of methylamine hydrochloride, 2.9 gm of paraformaldahyde, 0.31 gm of concentrated hydrochloric acid and 20 ml of isopropanol were added and this mixture was heated to reflux and stirred for 6 hrs. The mixture was then cooled to 0 °C and stirred for one hour more. The slurry was then filtered, and the solid was washed with cold ethanol. The washed solid was dried for 16 hrs at 50 °C to obtain 12.5 gm of 2-thienyl 2-methylaminoethyl ketone hydrochloride, as a white sold. A 12.0 gm portion of that intermediate product was stirred in 40 ml of ethanol at ambient temperature, and the pH of the solution was raised to 11-12 by slow addition of sodium hydroxide. A 1.03 gm portion of sodium borohydride was added, and the mixture was stirred at ambient temperature for 4 hrs. Then 7.5 ml of acetone was added, and the mixture was stirred for 20 minutes more. The mixture was then concentrated by evaporation to a white slurry, and 120 ml of methyl t-butyl ether was added. The mixture was acidified to pH 1-1.5 by addition of concentrated hydrochloric acid, and the solution was stirred for ten minutes. The pH was then made basic to pH 12 by slow addition of sodium hydroxide. The layers were then separated, the aqueous phase was extracted with 30 ml of methyl t-butyl ether and the organic phases were combined and washed once with 50 ml of water. The organic phase was concentrated by evaporation to get a solid product. The retro-synthetic pathway is shown in Figure 5. |
| |
| White solid, Yield 87%, 24.35 g, mp 72-73°C; IR (KBr) cm-1: 2824.49 (m, CH3-N st), 1654.25 (s, CH3N-H st as),1465.72 (m, C-H st as,-CH2-)1300.59 (s, C-O st), 689.25 (s, C-S st); 1H NMR (400 MHz, CDCl3): δ 7.21-7.28 (m, 1H, Ar-H), 6.93-7.01 (m,1H, Ar-H), 6.52-6.87 (m,1H, Ar-H), 2.79-3.02 (m, 2H, -CH2-CH2-NH-), 2.38 (d, 3H,-CH3), 3.50 (br, 2H, -CH2-CH2-NH), 1.18-2.12 (m, 1H, -NH), 3.64 (d,1H,- OH); 13C NMR (100 MHz, CDCl3):149.6, 126.4, 123.5, 122.1,71.9, 50.1, 36.8, 35.91; Mass (m/z) ESI TOF: 141.03 (C7H9OS), 172.07 (M+H), 173.07 (M+2H), 194.05 (M+Na), 210.06 (M+K). |
| |
| Chromatographic conditions |
| |
| The experiment was performed on a Bondapak C18 (250mm ×4.6mm, 5μm particle size) column using methanol-phosphate buffer (pH 7.8; 50 mM) (7:3, v/v) as the mobile phase at a flow rate of 1.5 ml/ min. The mobile phase was filtered through a nylon membrane filter paper (pore size 0.45 μm) and degassed with a sonicator for 10 min. The chromatography was performed at room temperature at a flow rate of 1.5 ml/min. The column temperature was maintained at 25 °C and eluents were monitored at a wavelength of 221 nm. The volume of each injection was 20 μl. |
| |
| Sample preparation |
| |
| The standard and sample stock solutions of duloxetine were prepared separately in a solvent mixture of methanol-phosphate buffer (pH 7.8; 50 mM) (7:3, v/v) at a concentration of 1000 μg/ml.Working standard solutions were prepared by diluting the standard stock solution with above solvent to get solutions of concentration in the range of 5-200 μg/ml. A stock solution of degradation product-1, 2 and 3 (500 μg/ml) were also prepared separately in mobile phase. |
| |
| Degradation studies |
| |
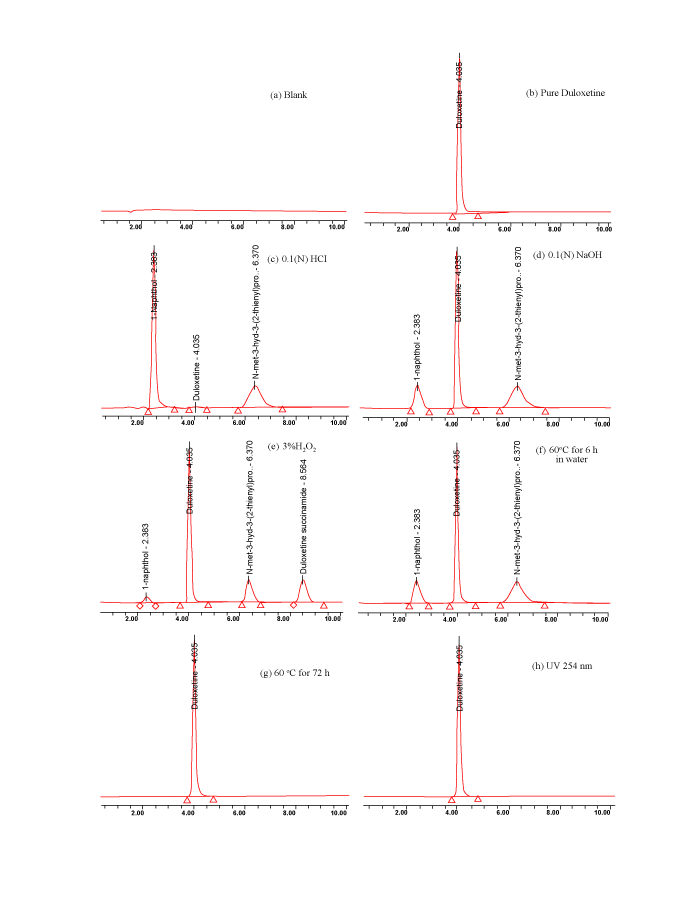
| All the degradation studies were carried out with drug solution of 100 μg/ml concentration. For acidic and alkaline hydrolysis studies, the drug was mixed with 0.1(N) HCl and 0.1(N) NaOH separately. These mixtures were refluxed on a water bath for 4 h at 60oC. The forced degradation in acidic and basic media was performed in the dark in order to exclude the possible degradative effect of light. The resulting solutions were neutralized by base and acid, respectively to avoid any interference of acid or base in the following steps. 20 μl of resulting solutions were injected into HPLC system and the chromatograms were recorded. About 89.5% and 3.5% degradation of drug were observed for acid and basic media, respectively. Among acid hydrolysis products, degradation product-1 was (about 46.69 %) observed at RT 2.383 min and degradation product-2 was (about 37.79 %) observed at RT 6.370 min. In basic hydrolysis, degradation product-1 was (about 2.0%) observed at RT 2.383 min and degradation product-2 was (about 1.5%) observed at RT 6.370 min. When duloxetine was subjected to oxidative degradation by treating with 3% hydrogen peroxide solution for 24 h at ambient temperature, the extent of degradation was about 12.52 %, out of which 2.71% corresponded to degradation product-1 (RT 2.383 min), 4.61 % was degradation product-2 (RT 6.370 min) and 5.2 % was degradation product-3 (RT 8.564 min). The aqueous solution of duloxetine refluxed for 6 h on a water bath set at 60 oC for wet heat degradation study, the extent of degradation was about 4.6 %, out of which 2.67 % corresponded to degradation product-1 (RT 2.383 min) and 1.93 % was degradation product-2 (RT 6.370 min). The drug product was also stored in incubator at 60 oC for 72h for dry heat degradation study and exposing to direct UV light (254 nm) for 24 h for photo chemical stability study. No degradation was observed in both the cases. In all cases, degradation product-1, 2 and 3 were confirmed by co-injection. |
| |
| Analysis of marketed formulation |
| |
| To determine the content of duloxetine in conventional tablets (label claim 20 mg/tablet), the tablets were powdered and the powder equivalent to 100 mg of ranolazine dihydrochloride was taken in a 100 ml volumetric flask and approximately 80 ml of suitable diluent [methanol- phosphate buffer (pH 7.8; 50 mM) (7:3, v/v)] was added to it. The content of the flask was then allowed to stand for 15min with intermittent sonication to ensure that the drug was completely dissolved and then filtered using 0.45 μm nylon membrane filter (Whatman). Final volume was made up to 100 ml with the above diluent to prepare a stock solution of 1000 μg/ml. 1.0 ml of the resulting solution was transferred into a 10 ml volumetric flask and the volume was made up to the mark with diluent. A sample of 20 μl of the resulting solution was directly injected. The average drug content of the formulations was determined from the corresponding regression equation. |
| |
| Method validation |
| |
| The developed chromatographic method was validated by evaluating the following analytical parameters: specificity, Precision, Limit of detection (LOD), Limit of quantification (LOQ), Accuracy, Linearity and Robustness. |
| |
| Specificity: Specificity is the ability of the method to measure the analyte response in the presence of its potential degradation products. Testing of the drug substance under stress conditions can help to identify the likely degradation products, which can in turn help to establish the degradation pathways and the intrinsic stability of the molecule. |
| |
| The specificity of the developed HPLC method for duloxetine was determined in the presence of its degradation products, namely degradation product-1, 2 and 3. Degradation was attempted under stress conditions like acid (0.1N HCl at 60oC for 4 h), base (0.1N NaOH at 60 oC for 4 h), oxidation (3% H2O2 at ambient temperature for 24 h), dry heat (60 oC for 72 h), wet heat (60 oC for 6 h) and UV light (254 nm for 24 h) to evaluate the ability of the proposed method to separate duloxetine from its degradation products. Peak purity test was performed for duloxetine peak by using tunable absorbance UV Visible detector in samples exposed to stress conditions. Assay studies were performed for stress samples against qualified duloxetine reference standard. Assay was also made for duloxetine bulk sample by spiking all the degradation products (1, 2 and 3) at the specification level (i.e., 0.15 %). |
| |
| Precision |
| |
| Intra Assay Precision: Intra assay precision was evaluated by performing six independent assays of the duloxetine at three concentration levels 80, 100, and 120% of assay analyte concentration(100 μg/ml) i.e., 80,100 and 120 μg /ml on the same day against qualified reference standard and calculating the % CV of assay. |
| |
| The intra assay precision of the analytical method of related substances was checked by injecting six individual preparations of stress duloxetine (100 μg/ml) spiked with 0.06 %, 0.12 % and 0.18 % each of degradation product-1, 2 and 3 with respect to stress duloxetine analyte concentration on the same day and calculating the % CV of degradation product-1, 2 and 3. |
| |
| Inter Assay Precision: Inter assay precision was evaluated by performing six independent assay of the duloxetine at three concentration levels 80, 100 and 120% of assay analyte concentration (100 μg/ml) i.e., 80, 100 and 120 μg/ml for 6 days against qualified reference standard and calculating the % CV of assay. |
| |
| The inter assay precision of the analytical method of related substances was checked by injecting six individual preparations of stress ranolazine dihydrochloride (100 μg/ml) spiked with 0.06%, 0.12 % and 0.18 % each of degradation product-1, 2 and 3 with respect to stress duloxetine analyte concentration for 6 days and calculating the % CV of degradation product-1, 2 and 3. |
| |
| Limit of detection (LOD) and Limit of quantification (LOQ): The LOD and LOQ for duloxetine, degradation product-1, 2 and 3 were estimated at a signal-to-noise ratio of 3:1 and 10:1, respectively by injecting a series of dilute solutions with known concentration (ICH 1995). Precision study was also performed at the LOQ level by injecting six individual preparations of duloxetine, degradation product-1, 2 and 3 by calculating the % CV of the AUC. |
| |
| Accuracy: The accuracy of the assay method was evaluated at three concentration levels i.e. 40 μg/ml, 50 μg/ml and 60 μg/ml in bulk drug sample. The percentage of recoveries was calculated from the slope and Y-intercept of the calibration curve obtained during linearity study of assay method. Accuracy/recovery experiment was repeated six times |
| |
| Standard addition and recovery experiments were conducted to determine accuracy of the related substance method for the quantification of degradation product-1, 2 and 3 in duloxetine stress samples. |
| |
| The study was conducted six times at 40%, 80% and 120% of specific level concentration. The percentage of recoveries for degradation product-1, 2 and 3 were calculated from slope and Y- intercept of the calibration curve obtained. |
| |
| Linearity: Linearity test solutions for assay method were prepared from stock solution at eleven concentration levels starting from 5 to 200 μg/ml. The peak area versus concentration data was performed by least-squares linear regression analysis. |
| |
| Linearity test solutions for related substance method were prepared by diluting stock solution to the required concentrations. The solutions were prepared at eleven concentration levels from LOQ to 200 % of the specification level. |
| |
| Linearity test was performed for three consecutive days in same concentration range for both assay and related substance method. The % RSD, value of the slope and Y-intercept of the calibration curve were calculated. |
| |
| Robustness: To determine the robustness of the developed method, experimental conditions were purposely altered and the resolution between duloxetine and degradation products was evaluated. |
| |
| The flow rate of the mobile was 1.5 ml/min. To study the effect of flow rate on the resolution, it was changed by 0.2 units from 1.3 to 1.5 ml/min. The effect of the column temperature on resolution was studied at 20 oC and 30 oC instead of 25 oC. The effect of the present organic strength on resolution was studied by varying methanol by -3 to +3 % while other mobile phase components were held constant. |
| |
| Solution stability and mobile phase stability |
| |
| The solution stability of duloxetine in the assay method was performed by leaving both the test solutions of the sample and reference standard in tightly capped volumetric flasks at room temperature for 48 h. The same sample solutions were analyzed for 6 h interval up to the study period. Furthermore, mobile phase stability was also performed by analyzing the sample solutions against freshly prepared reference standard solution for 6 h interval up to 48 h. Mobile phase prepared was kept constant during the study period. The % RSD for the assay of duloxetine was calculated during mobile phase and solution stability experiments. |
| |
| The solution stability of duloxetine and its degradation products in the related substance method were performed by leaving spiked sample solution in tightly capped volumetric flask at room temperature for 48 h. Content of degradation products 1, 2 and 3 were determined for every 6 h interval up to the study period. Mobile phase stability was also performed for 48 h by injecting the freshly prepared sample solutions for every 6 h interval. |
| |
| Result and Discussion |
| |
| Optimization of chromatographic conditions |
| |
| The main target of the chromatographic method is to get the separation of critical closely eluting degradation products, namely degradation product-1, 2 and 3. Degradation products were co eluted by using different stationary phases like C18, C8, phenyl and cyano and different mobile phases. The chromatographic separation was achieved on a Bondapak C18 (250 mm × 4.6 mm, 5 μm) column using methanolphosphate buffer (pH 7.8; 50 mM) (7:3, v/v) as a mobile phase. The flow rate of the mobile phase was 1.5 ml/min, at 25oC column temperature, the peak shape of duloxetine was found symmetrical. In optimized chromatographic conditions duloxetine, degradation product-1, 2 and 3 were well separated with resolution greater than 2, typical retention times were shown in Figure 6(e). The developed HPLC method was found to be specific for duloxetine and its degradation products. |
| |
| Results of forced degradation studies |
| |
| |
| Duloxetine was degraded in all conditions except dry heat and light (Figure 6). Peak purity test results confirmed that the duloxetine peak is homogenous and pure in all the analyzed stress samples. The assay of duloxetine is unaffected in the presence of its degradation products, confirms the stability indicating power of the developed method. The summary of forced degradation studies is given in Table 1. |
| |
|
|
Figure 6: Representative HPLC chromatograms of duloxetine pure and stressed samples. |
|
| |
|
|
Table 1: Summary report of forced degradation study. |
|
| |
| Precision |
| |
| The intra and inter assay coefficient of variation for assay of duloxetine during assay method precision study were within 0.33 and 0.42%, respectively (Table 2). |
| |
|
|
Table 2: Intra and Inter assay precision of duloxetine. |
|
| |
| The intra assay and inter assay coefficient of variation for degradation product-1,2 and 3 in related substance method precision study were within 0.48 and 0.56%, respectively (Table 3), conforming good precision of the method. |
| |
|
|
Table 3: Intra and Inter assay precision of degradation products. |
|
| |
| Limit of Detection and Limit of Quantification |
| |
| The limit of detection and limit of quantification of duloxetine was 0.081 μg/ml and 0.246 μg/ml respectively for 20 μl injection volume. The coefficient of variation of duloxetine at LOQ concentration was below 2.1%. |
| |
| The limit of detection of degradation product-1, 2 and 3 were 0.043, 0.051 and 0.037 μg/ml respectively for 20 μl injection volume. The limit of quantification of degradation product-1, 2 and 3 were 0.12, 0.18 and 0.13 μg/ml respectively for 20 μl injection volume. The coefficient of variation at LOQ concentration for degradation product-1, 2 and 3 were below 0.6%. |
| |
| Accuracy |
| |
| The percentage recovery of duloxetine in formulation samples was ranged from 99.05 to 99.97 (Table 4). |
| |
|
|
Table 4: Recovery result of duloxetine. |
|
| |
| The percentage recovery of degradation product-1 in stress sample was ranged from 99.99 to 101.1, for degradation product-2 in stress sample was ranged from 99.1 to 101.8 and for degradation product-3 in stress sample was ranged from 100.4 to 101.2 (Table 5). |
| |
|
|
Table 5: Recovery data for degradation products. |
|
| |
| Linearity |
| |
| Linearity calibration plot for assay method of duloxetine was obtained over the calibration ranges tested, i.e., 5-200 μg/ml .The average area under curve were plotted versus the concentrations injected. The equation for the resultant calibration curve was y =87697x + 28148 and correlation coefficient obtained was greater than 0.999. Linearity was checked for assay method over the same concentration range for three consecutive days. The % RSD values of peak areas were within 0.11 and 0.48%. The result shows that an excellent correlation existed between the peak areas and concentrations of analyte. |
| |
| Linear calibration plot for related substances method was obtained over the calibration ranges tested i.e. LOQ to 200% of the specification level for degradation product-1, 2 and 3.The correlation coefficient obtained was greater than 0.998. Linearity was checked for related substance method over the same concentration range for 3 consecutive days. The % RSD values of peak areas were within 0.16 to 0.47%. The result shows an excellent correlation existed between the peak areas and concentration of degradation product-1, 2 and 3. |
| |
| Robustness |
| |
| In all the deliberately varied chromatographic conditions performed (flow rate, column temperature and organic solvent), the resolution of degradation products in the stress sample mixture and of duloxetine remains unchanged, indicating that the analytical method was appropriately robust. |
| |
| Solution stability and mobile phase stability |
| |
| The % RSD of assay of of duloxetine during solution stability experiment was within 1.2%. No significant changes were observed in the content of degradation product-1, 2 and 3 during solution stability and mobile phase stability experiments when performed using related substances method. The solution stability and mobile phase stability experiments data confirm that sample solutions and mobile phase used during assay and related substance determination were stable up to 48hrs. |
| |
| Assay of of duloxetine marketed formulation |
| |
| The assay of Dulojoy and Dureep tablets containing of duloxetine was found to be 99.95% and 99.78% (Table 6). |
| |
|
|
Table 6: Assay of duloxetine in dosage form. |
|
| |
| Conclusion |
| |
| The reverse phase high performance liquid chromatography (HPLC) method was developed for quantitative and related substance determination of duloxetine is precise, accurate, rapid and specific. Acid and basic hydrolysis of duloxetine yielded two degradation products they were characterized as 1-naphthol and N-methyl-3- hydroxy-3-(2-thienyl) propylamine. Oxidative degradation reveals that duloxetine is degraded to 1-naphthol, N-methyl-3-hydroxy-3-(2- thienyl) propylamine and duloxetine succinamide. Like acid and basic hydrolysis wet heat also shows similar type of the degraded products. The method was completely validated showing satisfactory data for all the method validation parameters. The developed method can be used for the routine analysis and also to check the stability of duloxetine. |
| |
| Acknowledgement |
| |
| One of the authors (Tapas Kumar Laha) is grateful to University Grants Commission (UGC) for his minor research project grant, whose partial work is reported in here. Above all, the authors would like to offer their gratitude to the authorities of college of pharmaceutical sciences, Mohuda for providing all the facilities required to accomplish the work. |
| |
| |
| References |
| |
- The Merck Index (2006) An Encyclopedia of Chemicals, Drugs, and Biologicals (14th edn.) Merck and Co. Inc., Whitehouse Station, New Jersey, USA.
- Dasari S, Viriyala RK, Santosh K, Dnss AK, Bishat SPS, et al. (2010) A validated RP-HPLC method for the analysis of duloxetine hydrochloride in pharmaceutical dosage forms. Pharmacie Globale (IJCP) 3: 1-3.
- Mishra L (2006) Duloxetine hydrochloride is a newer selective serotonin and norepinephrine reuptake inhibitor (SSNRI) used for major depressive disorders. Drugs today 1: 489.
- Stephan AC, Luc AG, Francois RV, Peter RB, Frank PB, et al. (2003) Duloxetine increases serotonin and norepinephrine availability in healthy subjects: A double-blind controlled study. J Neuropsychopharmacolgy 28: 1685-1693.
- National Institute for Health and Clinical Excellence (2006) Urinary incontinence the management of urinary incontinence in women. London.
- National Institute for Health and Clinical Excellence (2010) Neuropathic Pain: The Pharmacological Management of Neuropathic Pain in Adults in Non-Specialist Settings. London.
- https://www.drugbank.ca/drugs/DB00476
- Wolfe F, Smythe HA, Yunus MB, Bennett RM, Bombardier C (1990) Criteria for the Classification of Fibromyalgia. Arthritis Rheum 33: 160–172.
- Douglas DR (2006) Treating Patients for Comorbid Depression, Anxiety Disorders, and Somatic Illnesses. J Am Osteopath Assoc 106: S1-S8.
- https://www.cancer.gov/dictionary/CdrID=589399
- Johnson JT, Oldham SW, Lantz RJ, Delong AF (1996) High performance liquid chromatographic method for the determination of duloxetine and desmethyl duloxetine in human plasma. J Liq Chromatogr Rel Technol 19: 1631–1641.
- Soni P, Mariappan TT, Banerjee UC (2005) High-performance liquid chromatographic method for the simultaneous estimation of the key intermediates of duloxetine. Talanta 67: 975–978.
- Brenna E, Frigoli S, Fronza G, Fuganti C, Malpezzi L (2007) Isolation and characterization of a phenolic impurity in a commercial sample of duloxetine. J Pharm Biomed Anal 43: 1573–1575.
- Jansen PJ, Oren PL, Kemp CA, Maple SR, Baertschi SW (1998) Characterization of impurities formed by interaction of duloxetine HCl with enteric polymers hydroxypropyl methylcellulose acetate succinate and hydroxypropyl methylcellulose phthalate. J Pharm Sci 87: 81–85.
- Kamila MM, Mondal N, Ghosh LK (2007) A validated UV spectrophotometric method for determination of duloxetine hydrochloride. Pharmazie 62: 414–415.
- Olsen BA, Argentine MD (1996) HPLC method development for duloxetine hydrochloride using a combination of computer-based solvent strength optimization and solvent selectivity mixture design. J Liq Chromatogr Rel Technol 19: 1993–2007.
- Dhaneshwar SS, Deshpande P, Patil M , Vadnerkar G, Dhaneshwar SR (2008) Development and validation of a HPTLC method for estimation of duloxetine hydrochloride in bulk drug and in tablet dosage form. Indian J Pharm Sci 70: 233–236.
- Musenga A, Amore M, Mandrioli R, Kenndler E, de Martino, et al. (2009) Determination of duloxetine in human plasma by capillary electrophoresis with laser-induced fluorescence detection. J Chromatogr B 877: 1126–1132.
- Patel SK, Patel NJ, Prajapati AM, Patel DB, Patel SA (2010) Stability-indicating RP-HPLC Method development and validation for Duloxetine Hydrochloride in Tablets. J AOAC Int 93: 123–132.
- Sinha VR, Kumria R, Bhinge JR (2009) Stress degradation studies on duloxetine hydrochloride and development of an RP-HPLC method for its determination in capsule formulation. J Chromatogr Sci 47: 589-593.
- Ramana NVVSS, Harikrishnaa KA, Prasada AVSS, Reddya KR, Ramakrishna K (2010) Determination of duloxetine hydrochloride in the presence of process and degradation impurities by a validated stability-indicating RP-LC method. J Pharm Biomed Anal 51: 994–997.
- Chhalotiya UK, Bhatt KK, Shah DA, Baldania SL (2010) Development and validation of a stability-indicating RP-HPLC method for duloxetine hydrochloride in its bulk and tablet dosage form. Sci Pharm 78: 857–868.
- Reddy PB (2009) Validation and stability indicating reverse phase-high performance liquid chromatography for the determination of duloxetine in tablets. Int J Chem Tech Res 1: 602-605.
- Reddy PRM, Sreeramulu1 J, Naidu PY, Reddy AR (2010) Stability indicating fast LC for the Simultaneous estimation of Intermediates and degradants of duloxetine hydrochloride. Chromatographia 71: 95–100.
|
| |
| |