| Research Article |
Open Access |
|
| Hiroshi Saito* |
| Department of Internal Medicine, Kawamura Hospital, Japan |
| *Corresponding author: |
Hiroshi Saito
Department of Internal Medicine
Kawamura Hospital, Japan
Tel: 052-831-4062
E-mail: eise@beetle.ocn.ne.jp |
|
| |
| Received August 31, 2012; Published September 08, 2012 |
| |
| Citation: Saito H (2012) Storage Iron Metabolism. 1:377. doi:10.4172/scientificreports. 377 |
| |
| Copyright: © 2012 Saito H. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| Evidences from studies on the storage iron metabolism are presented with reference to the items; the methods for determining iron stores, the relationships between storage iron and erythropoiesis, the relationship between storage iron and iron absorption, including the pathways of ferritin and hemosiderin iron in iron deposition and mobilization. In addition, the characteristic phases of increase and decrease of ferritin and hemosiderin iron measured by serum ferritin kinetics are presented. |
| |
| Keywords |
| |
| Serum ferritin; Storage iron metabolism; Determination of ferritin and hemosiderin iron; Pathway of ferritin and hemosiderin iron; Relationships between storage iron; Erythropoiesis and hepcidin |
| |
| Introduction |
| |
| Iron is an essential element for the living body. The human body stores iron mostly in liver, spleen, marrow and skeletal muscle in the form of ferritin and hemosiderin. Hemosiderin has been known as yellowish granules that can be stained by Prussian blue in the tissue cells. On the other hand, ferritin is invisible by photomicroscopy or may be faintly visible and stained diffusely in the tissue cells by Prussian blue, if concentrated. Ferritin and hemosiderin are iron containing proteins with magnetic susceptibility. Ferritin is water-soluble and heat-resistant up to 80°C, but hemosiderin is water-insoluble and thermally denatured. The total amount of body iron stores is around 600 to 1000 mg in the normal adult male and around 200 to 300 mg in the normal adult female [1]. The ratio of iron deficiency anemia in the menstruating female is less than 10% [2-4], and that of iron deficiency without anemia is around 20 to 40% in the menstruating female [3,4]. The amount of storage iron in the normal female increases gradually after menopause, but it is still lower [2] than the level of the normal male even after 20 years [5]. |
| |
| In a negative iron balance, reserved iron will be exhausted sooner or later, and results in iron deficiency. On the other hand in a positive iron balance, iron will be accumulated in the body and results in iron overload caused by the increase of iron absorption or blood transfusion or mistreatment. |
| |
| Storage iron behaves as if resisting the change in the iron density gradient [6,7]. This is a homeostatic tendency of the storage iron metabolism. |
| |
| Iron produces hazardous free radicals, those causing various disorders not only in iron overload, but also in localized iron deposition [ 8-12]. The transformation of ferritin into hemosiderin [13,14] might be the second best evolutionary step to reduce iron toxicity, compensating for the lack of iron excretion function of the human body. An iron chelating agent, deferasirox [15], with iron removing efficacy comparable to that of phlebotomy [7] is now in use for the treatment of transfusional iron overload. |
| |
| Knowledge of the storage iron metabolism seems essential not only for understanding the basis of the iron metabolism, but also for studies of the vast field of medicine. |
| |
| Clinical Methods For Determining Iron Stores |
| |
| Quantitative determination of iron stores |
| |
| Total amount of iron in the blood removed by phlebotomy [11,16-19] or that in the transfused blood can be determined by using the ratio of hemoglobin iron to hemoglobin as 0.0034. The total amount of iron stored after intravenous iron injection to patients with iron deficiency anemia can be obtained by subtracting the iron fixed in the increased hemoglobin in red cell mass, from the total amount of iron injected [ 20]. |
| |
| Semiquantitative estimation of iron stores |
| |
| The correlation between the total amount of storage iron and hemosiderin grain counts in biopsy sample has been used for the estimation of iron stores. |
| |
| Body surface monitoring methods such as dual-energy X-ray CT [ 21], super conduction quantum interference device susceptometry and Magnetic Resonance Imaging (MRI) [22] were introduced. However, other than for MRI, these methods are not adopted clinically. Although MRI is available for the detection of localized iron deposition, it has limitations for the quantitation of storage iron, in iron overload [7,23]. |
| |
| The development of the serum ferritin assay method by Addison et al. [24], achieved a break-through in studies on the storage iron metabolism. Serum ferritin assay enabled us to differentiate the hypochromic hypoferremic Anemia of Chronic Disease (ACD), such as inflammation anemia from iron deficiency anemia [24-27]. |
| |
| Serum ferritin may render a value higher than the actual storage iron level in patients with various inflammations, malignancies, hereditary hyperferritinemia-cataract syndrome. Therefore, appropriate examinations are needed for excluding such suspected cases of overestimation. Despite such disadvantages, serum ferritin has been evaluated highly for the diagnosis and treatment of patients with iron deficiency anemia and iron overload [2-7,24-27]. |
| |
| According to the report by Addison et al. [24], it suggested that the serum ferritin concentration might reflect the iron stores of the body, a rate [27] and a formula [2] were proposed for the conversion from serum ferritin into iron stores. However, such conversion methods do not always reflect the amount of iron stores because serum ferritin cannot reflect hemosiderin iron. |
| |
| Determination of ferritin and hemosiderin Iron |
| |
| Saito et al. [7] developed a clinical method for the simultaneous determination of ferritin and hemosiderin iron, by using a serum ferritin decrease curve, measured in the course of iron removal by phlebotomy and iron chelating. The method is based on the fact that the serum ferritin decrease curve is composed of the sum of two components, [28] a decreasing and recovering component. The decreasing component reflects the decrease in pre-existed tissue ferritin iron, and the recovering component reflects the increase of the tissue ferritin iron by removal of iron from hemosiderin, i.e. decreasing hemosiderin iron. |
| |
| Storage Iron and Erythropoiesis |
| |
| Human body reserves iron probably because the supply of a sufficient amount of iron is difficult by iron absorption, when there is an urgent need for erythropoiesis, as in the case of a large amount of blood loss. However, the mobilization of iron from iron stores cannot be increased fast enough to meet the acute need for iron, if erythropoiesis is elevated up to 6 to 8 times the normal [29]. |
| |
| The shortage of iron supply for increased erythropoiesis from storage results in hypoferremia, which causes iron-restricted erythropoiesis [ 30]. Iron deficiency anemia limits hemoglobin synthesis [31]. This is applied for the suppression of erythropoiesis in polycythemia vera. Hypoferremia also occurs after the supplementation of elements such as vitamin B12, folate and erythropoietin, in element deficiency anemia. The hypoferremia in ACD is caused by the inhibition of iron release from macrophages, under the control of the hepcidin-ferroportin system [ 32,33]. ACD may also be induced by the inhibition of erythropoiesis by hepcidin [34]. Mild anemia is observed often in the iron overload that disappears after iron removal therapy. Increased ineffective erythropoiesis accelerates iron absorption and results in iron overload in thalassemia. Iron overload occurs by increased iron absorption in hereditary atransferrinemia, despite the lack of transferrin. Generally, in non-iron deficiency anemia, hemoglobin iron kept in the decreased red cell mass, shifts to storage. |
| |
| Pollycove et al. [35] demonstrated the long lasting radioiron reflux from a miscible storage iron pool, in hereditary hemochromatosis. Such a radioiron reflux does not appear in patients with aplastic anemia, without erythropoiesis. The percent radioiron utilization at 14 days becomes higher than the percentage of iron in the red cell mass, because it takes at least 3 red cell life spans for the complete mixing of radioiron with pre-existing iron in the body of normal subjects [36]. Therefore, the mixing is accelerated in hemolytic anemia. Haptoglobin-bound hemoglobin and hemopexin-bound heme are cleared from circulation, stored and reutilized in intravascular hemolysis. |
| |
| |
| Hemosiderin appears in the erythroblasts in sideroblastic anemia, with ineffective erythropoiesis. Bessis et al. [37] observed erythroblasts surrounded by ferritin, using electron microscope. They speculated that such ferritin was supplied from marrow macrophages to erythroblast, for hemoglobin synthesis [37]. However, it seems difficult to confirm the direction of ferritin movement from marrow macrophages to erythroblast, morphologically. Cazzola et al. [38] and others assayed ferritin in erythrocytes. The problems of erythrocyte ferritin assay are its time consuming process and the difficulty of complete removal of cells, other than mature erythrocytes. On the other hand, Bessis et al. [37] reported that ferritin disappeared in the mature erythrocytes by electron microscopy. Zail et al. [39] also reported that ferritin that existed in erythrocyte precursors, was lost in circulating mature erythrocytes. |
| |
| Storage Iron And Iron Absorption |
| |
| In 1948, Granick [40] proposed the mucosal block theory, which implied an automatic control of iron absorption by the saturation of ferritin formation in the mucosal epithelial cells, following oral iron administration. Saito [41] reported that hemosiderin formation was not saturated, although ferritin formation was saturated after oral iron administration in rats. He then thought that the blockage of iron absorption by way of hemosiderin formation might be incomplete [41]. The above-mentioned findings would seem to indicate a difference in the ferritin and hemosiderin iron metabolism, as discussed later. |
| |
| Iron can enter the intestinal epithelial cells from both the blood circulation and intestine. It becomes intracellular labile iron [42] and then enters the blood circulation, or is synthesized into ferritin and hemosiderin there or lost [43-45]. The stored iron leaves intestinal epithelial cells for blood circulation sooner or later in a negative iron balance, if the iron is not lost by the exfoliation of intestinal epithelial cells. |
| |
| Gene mutation causes iron overload by uncontrolled increase of iron absorption in hereditary hemochromatosis [27,32,46]. However, the relationship between gene mutation in hereditary hemochromatosis and hepcidin is not entirely clarified yet [32]. Iron absorption is not always increased in iron overloaded patients with hereditary hemochromatosis [46,47]. It is increased above normal in patients with hereditary hemochromatosis, whose iron storage is kept within the normal level after phlebotomy therapy [47]. Shiono et al. [48] revealed that the larger the iron stores before iron removal, the faster the rebound by the acceleration of iron absorption after phlebotomy therapy for patients with chronic hepatitis C [11]. |
| |
| Crosby [49] proposed a hypothesis on iron absorption that iron entering the epithelial cells was incorporated into a “ferritin apparatus” from which iron could not be released, but lost. However, his hypothesis seems unlikely because the formation and decomposition of ferritin and hemosiderin are rapid [7], thereby indicating the involvement of ferritin and hemosiderin iron in iron absorption, in a negative iron balance [7]. |
| |
| The storage iron level in the intestinal epithelial cells is controlled not only by hepcidin-ferroportin system [32,50], but also by erythropoiesis. Hepcidin production in the liver is controlled inversely by the storage iron level [32,51]. As mentioned above, iron absorption is controlled by interacting factors; storage iron, erythropoiesis and the hepcidinferroportin system. |
| |
| Intestinal iron absorption is the normal route of iron entering the body. Iron that enters it through abnormal routes by intravenous injection or red cell transfusion follows a different course from the iron absorbed via the normal route. |
| |
| Transferrin-bound iron after intravenous injection is taken up by the erythroid precursor cells, utilized for hemoglobin synthesis and fixed in the erythrocytes for the red cell life span, or remains unutilized in the form of ferritin and hemosiderin. The transfused effete red cell or intravenously injected colloidal iron is captured by the Reticulo Endothelial (RE) cells. There red cell or colloidal iron is decomposed and its iron is stored there once. Then, the stored iron in RE cells is released gradually and redistributed by transferrin to the tissues, in proportion to the ratio of pre-existing iron [28,52]. |
| |
| Pathways of Ferritin and Hemosiderin Iron |
| |
| Pathways of ferritin and hemosiderin iron in iron deposition |
| |
| Shoden et al. [28] proposed an iron pathway from plasma to ferritin, and from ferritin to hemosiderin in iron deposition. Their proposal seems to be supported by the transformation of ferritin into hemosiderin by various measures [14,15,28]. Shoden et al. [28] proposed an iron pathway from plasma to hemosiderin, glancing off ferritin in iron deposition. However, the nature of such a pathway seems unclear. |
| |
| Shoden et al. [28] also proposed an iron pathway from hemosiderin to ferritin in iron deposition. However, such a pathway seems unlikely, because its direction is contrary to the iron flow in iron deposition [7]. The same investigators also proposed a direct iron pathway from plasma to hemosiderin, bypassing ferritin synthesis in iron deposition [28]. However, such a pathway seems unlikely because intracellular labile iron will be involved in very active ferritin synthesis, as seen by the prompt serum ferritin increase after intravenous iron injection in patients with iron deficiency anemia. Furthermore, the detection of radioiron in hemosiderin fractions separated from the tissue homogenate soon after a radioiron addition, does not always indicate direct radioiron incorporation into hemosiderin, since it proved difficult to distinguish adhesion from incorporation [7] |
| |
| Thus, one iron pathway seems to exist in iron deposition, where iron flows in the numbered order of (1) hemosiderin → (2) ferritin→ (3) labile iron pool in iron deposition. |
| |
| Pathways of ferritin and hemosiderin iron in iron mobilization |
| |
| Shoden et al. [28] advanced an uncertain proposition regarding an iron pathway from hemosiderin to ferritin, and from ferritin to plasma in iron mobilization. This was confirmed by Saito et al. [7]. Shoden et al. [28] also proposed an unproven iron pathway from ferritin to hemosiderin, in iron mobilization. However, such a pathway seems unlikely, because its direction is contrary to the iron flow in iron mobilization. They [28] also proposed a direct iron pathway from hemosiderin to plasma, in iron mobilization. However, such a pathway seems unlikely because the iron split off from hemosiderin will be involved in active ferritin synthesis by removing iron from hemosiderin in iron mobilization, because the iron flows from ferritin to labile iron in iron mobilization, and because the recovery of labile iron pool utilizing ferritin iron in the negative iron balance is demonstrated [53]. |
| |
| Thus, one iron pathway seems to exist in iron mobilization [7], where iron decreases in the numbered order of (1) hemosiderin ← (2) ferritin ← (3) labile iron pool in iron mobilization, tracing the same route to iron deposition. |
| |
| Phases of increase and decrease of ferritin and hemosiderin iron |
| |
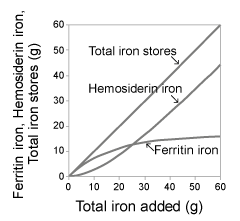
| The increase of ferritin iron was larger than that of hemosiderin iron initially, but it was smaller later. In contrast, the increase of hemosiderin iron was smaller than that of ferritin iron initially, but it was larger later, in the course of linear increase of total iron stores in a constant amount of iron addition (Figure 1). The saturation of ferritin iron increase reflects the transformation of ferritin into hemosiderin [ 13,14] in higher level of iron stores. The saturation indicates the limitation of the iron storing capacity of ferritin [7]. The linear increase of hemosiderin [7,28] implies the limitlessness of iron storing capacity of hemosiderin [7]. |
| |
|
|
Figure 1: (left) shows the phases of increase of ferritin iron, hemosiderin iron and total iron stores in the course of blood transfusion for a patient with aplastic anemia. |
|
| |
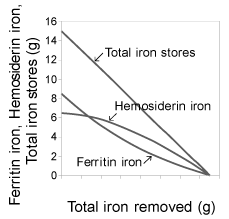
| The decrease of ferritin iron was larger than that of hemosiderin iron initially, but it was smaller later. In contrast, the decrease of hemosiderin iron was smaller than that of ferritin initially, but it was larger later in the course of the linear decrease of total iron stores, in a constant amount of iron removal (Figure 2). |
| |
|
|
Figure 2: (right) shows the phases of decrease of pre-existed ferritin iron (8.5 g), hemosiderin iron (6.5 g) and total iron stores (15 g) in the course of phlebotomy for a patient with hereditary hemochromatosis. |
|
| |
| |
| References |
| |
- Bothwell TH (1979) Iron Metabolism in Man. Blackwell Scientific Publications, Oxford, United kingdom.
- Cook JD, Skikne BS, Lynch SR, Reusser ME (1986) Estimates of iron sufficiency in the US population. Blood 68: 726-731.
- Cook JD, Finch CA, Smith NJ (1976) Evaluation of the iron status of a population. Blood 48: 449-455.
- Uchida T (2004) Anemia in Japanese women: the current situation and the cause. Rinsho Ketsueki 45: 1085-1089.
- Saito H, Ishikawa K (1985) Ferritin In: Nippon-Rinsho. Jpn J Clin Med 532: 122-125.
- Fleming RE, Bacon BR (2005) Orchestration of iron homeostasis. N Engl J Med 352: 1741-1744.
- Saito H, Tomita A, Ohashi H, Maeda H, Hayashi H, et al. (2012) Determination of ferritin and hemosiderin iron in patients with normal iron stores and iron overload by serum ferritin kinetics. Nagoya J Med Sci 74: 39-49.
- Eaton JW, Qian M (2002) Molecular bases of cellular iron toxicity. Free Radic Biol Med 32: 833-840
- Stevens RG, Jones DY, Micozzi MS, Taylor PR (1988) Body iron stores and the risk of cancer. N Engl J Med 319: 1047-1052.
- Sullivan JL (1989) The iron paradigm of ischemic heart disease. Am Heart J 117: 1177-1188.
- Hayashi H, Takikawa T, Nishimura N, Yano M, Isomura T, et al. (1994) Improvement of serum aminotransferase levels after phlebotomy in patients with chronic active hepatitis C and excess hepatic iron. Am J Gastroenterol 89: 986-988.
- Miyajima H, Nishimura Y, Mizoguchi K, Sakamoto M, Shimizu T, et al. (1987) Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology 37: 761-767.
- Matioli GT, Baker RF (1963) Denaturation of ferritin and its relationship with hemosiderin. J Ultrastruct Res 8: 477-490.
- Miyazaki E, Kato J, Kobune M, Okumura K, Sasaki K, et al. (2002) Denatured H-ferritin subunit is a major constituent of haemosiderin in the liver of patients with iron overload. Gut 50: 413-419
- Cappellini MD, Cohen A, Piga A, Bejaoui M, Perrotta S, et al. (2006) A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 107: 3455-3462.
- Haskins D, Stevens AR Jr, Finch SC, Finch CA (1952) Iron metabolism; iron stores in man as measured by phlebotomy. J Clin Invest 31: 543-547.
- Milder MS, Cook JD, Stray S, Finch CA (1980) Idiopathic hemochromatosis, an interim report. Medicine (Baltimore) 59: 34-49.
- Prieto J, Barry M, Sherlock S (1975) Serum ferritin in patients with iron overload and with acute and chronic liver diseases. Gastroenterology 68: 525-533.
- Van Oost BA, van den Beld B, van Ashbeck BS, Marx JJ (1985) Monitoring of intensive phlebotomy therapy in iron overload by serum ferritin assay. Am J Hematol 18: 7-12.
- Saito H, Maeda H (2004) Method for determining the amount of blood loss using the storage iron decrease rate as obtained from serum ferritin after intravenous iron therapy. Rinsho Ketsueki 45: 1177-1180.
- Goldberg HI, Cann CE, Moss AA, Ohto M, Brito A, et al. (1982) Noninvasive quantitation of liver iron in dogs with hemochromatosis using dual-energy CT scanning. Invest Radiol 17: 375-380
- Jensen PD, Jensen FT, Christensen T, Ellegaard J (1994) Non-invasive assessment of tissue iron overload in the liver by magnetic resonance imaging. Br J Haematol 87: 171-184.
- Angellucci E, Giovagnoni A, Valeri G, Paci E, Ripalti M, et al. (1997) Limitation of magnetic resonance imaging in measurement of hepatic iron. Blood 90: 4736-4742.
- Addison GM, Biemish MR, Hales CN, Hodgkins M, Jacobs A, et al. (1972) An immunoradiometric assay for ferritin in the serum of normal subjects and patients with iron deficiency and iron overload. J Clin Pathol 25: 326-329.
- Cook JD, Lipschitz DA, Miles LE, Finch CA (1974) Serum ferritin as a measure of iron stores in normal subjects. Am J Clin Nutr 27: 681-687.
- Jacobs A, Worwood M (1975) Ferritin in serum. Clinical and biological implications. New Engl J Med 292: 951-956.
- Walters GO, Miller FM, Worwood M (1973) Serum ferritin concentration and iron stores in normal subjects. J Clin Pathol 26: 770-772.
- Shoden A, Gabrio BW, Finch CA (1953) The relationship between ferritin and hemosiderin in rabbits and man. J Biol Chem 204: 823-830.
- Crosby WH (1958) Treatment of hemochromatosis by energetic phlebotomy; one patient’s response to the letting of 55 litres of blood in 11 months. Br J Haematol 4: 82-88.
- Hillman RS, Henderson PA (1969) Control of marrow production by the level of iron supply. J Clin Invest 48: 454-460.
- Finch S, Haskins D, Finch CA (1950) Iron metabolism; hematopoiesis following phlebotomy; iron as a limiting factor. J Clin Invest 29: 1078-1086.
- Ganz T (2003) Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 102: 783-788.
- Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, et al. (2004) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306: 2090-2093.
- Dallalio G, Law E, Means RT Jr (2006) Hepcidin inhibits in vitro erythroid colony formation at reduced erythropoietin concentrations. Blood 107: 2702-2704.
- Pollycove M, Mortimer R (1961) The quantitative determination of iron kinetics and hemoglobin synthesis in human subjects. J Clin Invest 40: 753-782.
- Finch CA (1959) Body iron exchange in man. J Clin Invest 38: 392-396
- Bessis MC, Breton-Gorius J (1962) Iron metabolism in the bone marrow as seen by electron microscopy: a critical review. Blood 19: 635-663.
- Cazzola M, Dezza L, Bergamaschi G, Barosi G, Bellotti V, et al. (1983) Biologic and clinical significance of red cell ferritin. Blood 62: 1078-1087.
- Zail SS, Charlton RW, Torrance JD, Bothwell TH (1964) Studies on the formation of ferritin in red cell precursors. J Clin Invest 43: 670-680.
- Granick S (1946) Ferritin; its properties and significance for iron metabolism. Chem Rev 38: 379-403.
- Saito H (1958) Studies on storage iron. Dynamic behavior of ferritin and hemosiderin under various experimental conditions. Nagoya J Med Sci 21: 288-300.
- Jacobs A (1977) Low molecular weight intracellular iron transport compounds. Blood 50: 433-439.
- Conrads ME Jr, Crosby WH (1963) Intestinal mucosal mechanisms controlling iron absorption. Blood 22: 406-415.
- Saito H, Sargent T 3rd, Parker HG, Lawrence JH (1964) Whole body iron loss in normal man measured with a gamma spectrometer. J Nucl Med 5: 571-580.
- Green R, Charlton RW, Seftel H, Bothwell T, Mayet F, et al. (1968) Body iron excretion in man: a collaborative study. Am J Med 45: 336-353
- Williams R, Manenti F, Williams HS, Pitcher CS (1966) Iron absorption in idiopathic haemochromatosis before, during, and after venesection therapy. Brit Med J 2: 78-81.
- Sargent T, Saito H, Winchell HS (1971) Iron absorption in hemochromatosis before and after phlebotomy therapy. J Nucl Med 12: 660-667.
- Shiono Y, Hayashi H, Wakusawa S, Sanae F, Takikawa T, et al. (2001) Body iron stores and iron restoration rate in Japanese patients with chronic hepatitis C as measured during therapeutic iron removal revealed neither increased body iron stores nor effects of C282Y and H63D mutations on iron indices. Nagoya J Med Sci 64 : 51-57.
- Crosby WH (1963) The control of iron balance by the intestinal mucosa. Blood 22: 441-449.
- Young MF, Glahn RP, Ariza-Nieto M, Inglis J, Olbina G, et al. (2009) Serum hepcidin is significantly associated with iron absorption from food and supplemental sources in healthy young women. Am J Clin Nutr 89: 533-538.
- Ganz T (2006) Hepcidin and its role in regulating systemic iron metabolism. Iron in Hematology. American Society of Hematology29-35.
- Gross F, Naegeli SR, Philips HD (1963) Iron metabolism: An international symposium sponsored by Ciba, Aix-en-Provence. (1st edition), Springer, Germany.
- Konijn AM, Glickstein H, Vaisman B, Meyron-Holze EG, Slotki IN, et al. (1999) The cellular labile iron pool and intracellular ferritin in K562 cells. Blood 94: 2128-2134.
|
| |
| |