| Research Article |
Open Access |
|
| Fiorenzo Mignini1*, Cinzia Nasuti1, Giorgio Gioventu1, Valerio Napolioni2, Roberto Fasani1 and Ferrari P1 |
| 1School of Farmacy and Health product, University of Camerino, Camerino, Italy |
| 2School of Biosciences and Biotechnologies, University of Camerino, Camerino, Italy |
| *Corresponding authors: |
Fiorenzo Mignini
Professor
School of Farmacy and Health Product Experimental Medicine Unit
University of Camerino
Via Madonna delle Carceri
Camerino (MC), Italy
Tel: +39 0737 403304
Fax: +39 0737 403325
E-mail: fiorenzo.mignini@unicam.it |
|
| |
| Received April 04, 2012; Published October 31, 2012 |
| |
| Citation: Mignini F, Nasuti C, Gioventu G, Napolioni V, Fasani R et al., (2012) Human Bioavailability and Pharmacokinetic Profile of Different Formulations Delivering Alpha Lipoic Acid. 1:418. doi:10.4172/scientificreports.418 |
| |
| Copyright: © 2012 Mignini F, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| |
| Abstract |
| |
| Alpha-lipoic acid (ALA) is an important micronutrient with several pharmacologic as well as antioxidant properties. Oral formulations present problems due to characteristics of the molecule and of the pharmaceutical forms. It is known that ALA is poorly soluble; therefore to increase the solubility was reticulated in an amphiphilic matrix like lecithin. The goal of the present study was to characterize the bioavailability of new formulation and to compare the human pharmacokinetics profiles of two different pharmaceutical form: tablets and soft gel capsules following single oral administration of a ALA 600 mg. Blood samples were collected up to 8 h post dosing, and plasma α-lipoic acid concentrations were determined by Liquid Chromatography Mass Spectrometry (LC/MS/MS) detection. The results revealed that after rapid dissolution there is a good solubilisation by lecithin and that the two formulations show the same human pharmacokinetic profile. Tablet formulation is more versatile than soft capsules because it makes it possible to administer 600 mg of ALA with good compliance. The properties of the new formulation ensure rapid release in vivo and good bioavailability with high ALA content per unit dose and excellent homogeneity of release. |
| |
| Keywords |
| |
| α-Lipoic acid; Bioavailability; Human pharmacokinetic; New oral formulation; Amphiphilic matrix; Surfactants; Disaggregating agents |
| |
| Abbreviations |
| |
| Cmax: maximum plasma concentration; AUCt: Area Under the plasma concentration-time Curve from time zero up to time t; Tmax: time of maximum plasma concentration |
| |
| Introduction |
| |
| Alpha-lipoic acid (1,2-dithiolane-3-valeric acid) (ALA), also known as thiotic acid, was discovered in 1937. Unlike antioxidant vitamins, ALA exhibits this activity in both the reduced and oxidated forms. Lipoic acid is a racemic mixture. The redox couple α-lipoic/dihydrolipoic acid containing the R-enantiomer is covalently bound to a lysine residue, forming an essential lipoamide, which functions as a co-enzyme for the E2 subunit of four multi-enzymatic mitochondrial complexes, for example pyruvate dehydrogenase [1-6]. After administration, ALA is reduced at the intracellular level by various enzymes and released into the extracellular environment as its principal metabolite, dihydrolipoic acid (DHLA) [7,8]. |
| |
| Oral formulations of α-lipoic acid present problems due to characteristics of the molecule (short blood half life, high presystemic elimination and hepatic first pass effect) and characteristics of the pharmaceutical forms. ALA is a poorly soluble molecule; the techniques used to increase its solubility in the gastrointestinal environment, such as micronisation and salification, have some drawbacks and disadvantages. Complexes and composites based on cyclodextrins or other polymers require costly processes that are often difficult to carry out and do not ensure complete complexation of the active ingredient. In addition, the active ingredient to polymer ratio is often a limiting factor in the preparation of an easy-to-administer pharmaceutical form. Micronisation processes do not always ensure significant increases in plasma levels, and the consequent increase in apparent density/ volumes and surface areas of the powders complicates the production of capsules, tablets and granulates. Emulsions and/or microemulsions, either simple or multiple, are often unstable and cannot carry pharmacologically active amounts of the medicament. Salification and/or solubilisation processes of conventional pharmaceutical forms sometime cannot improve the bioavailability of medicaments that are only slightly permeable and absorbable, or lipophilic ones, due to reprecipitation of the active ingredient in biological fluids, thus removing the advantage of a technological process to dissolve the medicament in the pharmaceutical formulation. |
| |
| A new oral formulation of ALA, called ALA 600, uses a patented technology (EP1401405 B 1) to deliver significantly bio available and safe quantities of α-lipoic acid to the bloodstream. An amphiphilic matrix with surfactants coupled with super disaggregating agent’s increases the dissolution rate in vitro with improved bioavailability and less variability in absorption. The compositions can be obtained with a process which comprises the following steps: 1) adding surfactants to the amphiphilic matrix, to obtain a homogeneous solution or dispersion (preferably in amounts from 0.1% to 5%); 2) solubilising, suspending, dispersing, totally or partly embedding one or more active principles (preferably from 0.1%. to 4.9%); 3) adding cyclodextrins and/or superdisgregatings agents; (preferably from 0.1% to 2.5%, to obtain a homogeneous dispersion); 4) optionally adding excipients; 5) optionally film-coaling with cellulose derivatives or methacrylic acid polymers. In a pilot study, we have observed that, respect to the recent published data, the new ALA formulation tends to show an improvement of Cmax and AUC values [9]. The goal of the present study was to characterize the bioavailability of new formulation and to compare the human pharmacokinetics profiles of two different pharmaceutical forms: tablets and soft gel capsules following single oral administration of an ALA 600 mg. |
| |
| Materials and Methods |
| |
| Pharmaceutical section |
| |
| Investigational products: Both test preparations, the nutritional supplements soft gel capsules (ALAnerv) 300 mg and tablets 600 mg (ALA600), will be supplied by the Alfa Wassermann. |
| |
| One soft gel capsule contains: active principles a-lipoic acid mg 300, polyunsaturated fatty acids (linoleic acid, α-linolenic acid) mg 180, natural vitamin E/d-alpha-tocopherol 7.5 mg, vitamins of the B complex (B1/thiamine monohydrate, 1.05 mg; B2/riboflavin, 1.20 mg; B5/calcium d-pantothenate, 4.5 mg; B6/pyridoxine chlorohydrate, 1.5 mg) and selenium methionine 25 mcg. Excipients: alimentary gelatine, glycerol, fatty acid triglycerides, magnesium stearate, polyglycerol oleate, soya oil, soya lecithin, titanium dioxyde, red iron oxide. |
| |
| One tablet of ALA600 contains: Active principles a-lipoic acid mg 600 and vitamins of the B complex (B1/thiamine monohydrate, 2.1 mg; B2/riboflavin, 2.4 mg; B5/calcium d-pantothenate, 9 mg; B6/ pyridoxine chlorohydrate, 3 mg). Excipients: di-calcium phosphate, cellulose microcrystalline, maltodextrins, polyvinylpyrrolidone, polyvinylpolypyrrolidone, colloidal silica, magnesium stearate, croscarmellous sodium, talcum powder, soya lecithin, hydroxypropylmethylcellulose, stearic acid, titanium dioxyde, red iron oxide. |
| |
| Pharmaceutical process |
| |
| The patent of oral formulation (EP 1401405 B1) was developed to improve the bioavailability of active ingredients added to the amphiphilic matrix, which are poorly absorbed via the oral route due to high variability of absorption in the gastrointestinal tract. This technology, which was originally used to deliver drug substances, has been applied in other fields, such as alimentary supply to obtain prompt-release and fast-absorption, and to enhance the bioavailability of lipoic acid. |
| |
| In the step 1) the amphiphilic matrix is prepared. Any amphiphilic semisolid excipients or mixtures thereof are melted above 60°C, or solubilised or suspended in solvent (preferably water) to obtain a homogeneous solution/dispersion, which becomes again semisolid or solid at room temperature with eutectic properties at temperatures ranging from 35°C to 37°C (body temperature) or able to be used as granulating system. Afterwards, said excipients, which have become liquid upon melting or are already liquid at room temperature, are added with surfactants to obtain a homogeneous dispersion. In step 2), the active ingredient is solubilised, dispersed and/or embedded in the surface-activated amphiphilic matrix from step 1) to obtain a homogeneous solution, or dispersion, or granules. In step 3), the system from step 2) is added with different amounts of cyclodextrins and/or superdisgregants until homogeneous dispersion. The resulting system can be distributed into soft- or hard-gelatine capsules to obtain a liquid, semisolid or solid pharmaceutical form inside the capsule. Alternatively, the system from step 2) can be loaded onto cyclodestrin and/or superdisgregrants and/or mixtures thereof to obtain powder, microgranules or granules having good free-flowing and/or lab letting characteristics. In step 4), excipients with different functions may be added to transform liquid or semisolid formulations into solid ones for the preparation of capsules, tablets, granulates, microgranules, minitablets, sachets, said excipients being, for example, silica, celluloses, starches, sugars, polyvinyl pyrrolidines, methacrylates, glidants, antiaggregants, lubricants such as magnesium stearate, stearic acid, talc, or the liquid semisolid formulations can be added with other liquid cosolubilizers such as, water, polyethylene glycols, glycerin, sorbitol. Amphiphilic compounds could be polar lipids (lecithin, phosphatidyl choline, phosphatidyl diethanolamine), glycol alkyl ethers such as dielhylene glycol monoethyl ether (Transcutol®). Surfactants comprise phosphatides and lecithins (phosphaiidyl cholines, phosphatidyl diethanolamines, sphyngomyelins), anionic and non-ionic emulsifying waxes, sodium lauryl sulfate, sodium dodecyl sulfate, polysorbales, cholic acids, poloxamer, sodium sulfosuccinate, sodium lauryl sarcosinate. Superdisgregrants comprise sodium starch glycolate (Explotab®), croscarmellose sodium (Acdisol®) crosslinked polyvinylpyrrolidone (Amberlites© (IRP SS). |
| |
| Pharmacokinetics section |
| |
| Ethics: The trial protocol, informed consent form and subject recruitment procedures were approved by the Joint Ethical Committee (JEC) Universita di Camerino-Azienda ASUR Marche ZT-10 of Camerino. Before signing the informed consent form the volunteer were informed in detail by a physician. The subject was given ample time to enquire about the details of the trial. The consent form was signed by the informing physician and by the volunteer. The study was conducted in accordance with the Declaration of Helsinki in its revised edition, the Guidelines of Good Clinical Practice (CPMP/ICH/135/95) and the Directives 2001/20/EC and 2005/28/EC and with international and local regulatory requirements. |
| |
| Subjects: demographic and other baseline characteristics: A sample size of 18 subjects was considered adequate to accurately assess the bioavailability and the pharmacokinetics parameters of the investigational drug. The 18 healthy volunteers were 9 males and 9 females. Their demographic and baseline characteristics are summarized in (Tables 1 and 2); the vital signs are summarized in ( Table 3). Three of the female volunteers have a Body Mass Index (BMI) lower than the limit 20, but they were healthy considering all the others parameters, so it was evaluated the ratio lean mass/fat mass by bioelectric impedance technique and they have values similar to the other volunteers, considering this they were enrolled. |
| |
|
|
Table 1: Demographic Data and Baseline Characteristics – Males. |
|
| |
|
|
Table 2: Demographic Data and Baseline Characteristics - Females. |
|
| |
|
|
Table 3: Vital signs (Minimum, Maximum, Mean and Standard Deviation)` |
|
| |
| Presence of cardiac, pulmonary, gastrointestinal, endocrine, musculoskeletal, neurological, haematological, hepatic or renal disease, unless deemed not clinically significant by the investigator; presence of any significant physical or organ abnormality; history or evidence of psychiatric or psychological disease (including depression) unless deemed not clinically significant by the investigator; any clinically significant illness during the 4 weeks before this study; pregnancy, lactation period; smoking; history of alcohol and drug abuse; known history of hypersensitivity to α-lipoic acid and to other sulphur molecules; use of any prescription medication in the 14 days preceding this study; use of over-the-counter (OTC), homeopathic and herbal medicines in the 14 days preceding this study; use of preparations containing α-lipoic acid in the 30 days preceding this study; participation in a clinical trial with an investigational drug in the 6 months preceding this study; blood donation in the month preceding this study; participation as a plasma donor in a plasmaphoresis programme in the 7 days preceding this study; subjects who are unable or unwilling to adhere to the protocol procedures; withdrawal of informed consent. |
| |
| Treatment: Softgel capsules ALA 2×300 mg (treatment A) and tablets ALA 1×600 mg (treatment B) was orally administered. Each subject received, in the fasted state, a single dose of the first randomly assigned treatment (A/B) in the morning of day 1, and, after 1 week wash-out at least, he/she received a single dose of the second randomly assigned treatment (A/B). |
| |
| Subjects were assigned to a treatment sequence (A/B or B/A) using a randomisation list (prepared by the Drug Safety Unit Officer of the Sponsor), which is known to the Drug Safety Unit Officer of the Sponsor only. It was opened at the end of analytical measurements, when pharmacokinetic and statistical analyses are to be performed and then archived in the Trial Master File. |
| |
| Pharmacokinetic Assessment |
| |
| Blood samples were taken at the following times: pre-dose (5 min before drug dosing); 5, 15, 30, 45 minutes and 1, 1.5, 2, 3, 4, 6 and 8 hours post-dose. Blood samples were collected in plain plastic heparinised tube and centrifuged within 20 minutes at 2000 rpm for 15 minutes. The plasma was divided into three aliquots of 1 ml. Plasma was shock frozen at -20°C within 1 hour from blood withdrawal and stored at -80°C until analysis. The following pharmacokinetic parameters of α-lipoic acid were calculated: the maximum plasma concentration (Cmax); time of maximum plasma concentration (Tmax); terminal half-life (t½:); Area Under the plasma concentration-time Curve from time zero up to time t (AUCt), where t is the last time point at which the subject showed concentrations above the lower limit of quantification i.e. time of last measurable (non-zero) concentration (t-last) using the trapezoidal rule; Area Under the plasma concentration-time Curve from time of dosing extrapolated to infinity using the trapezoidal rule (AUCinf). AUCs were computed using the Log Linear Method, trapezoidal when Cn>Cn-1. |
| |
| The study was carried out under quality assurance and quality control systems with written Standard Operating Procedures (SOP) in accordance with the Good Clinical Practice (GCP) Guidelines (CPMP/ ICH/135/95). Quality assurance was guaranteed by regular monitoring of the study by a qualified monitor. |
| |
| α-Lipoic Acid Assay: The operating procedures were carried out according to International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use. ICH harmonised tripartite guideline (Current Step 4 version. Complementary guideline on methodology dated 6 November 1996 incorporated in November 2005). |
| |
| ALA plasma levels were determined by high-performance liquid chromathography HPLC (Agilent 1200 SL) equiped with triple quadrupole mass detector (Agilent 6410) and column Acquity UPLC BEH C18, 50 mm × 2.1 mm × 1.7 μm. The mobile phase was made up of 5 mM of ammonium acetate solution for LC/MS (0.3854 g/L) in water. |
| |
| The stock solutions of lipoic acid (SSLA, Sigma 117K0679) and internal standard (IS) were prepared in methanol. The internal reference was cyclohexanobutyric acid (Aldrich, 00105LA). Calibration curves were prepared for both solutions by spiking each plasma blank samples with proper volume of the standard solutions to obtain the calibration curve points of ALA. The standard calibration curves for ALA were constructed using the analyte/IS peak area ratios versus the nominal concentrations of the analytes. Chromatographic separations were carried out using acetonitrile as mobile phase and acetic acid 0.1% (pH 4.0 adjusted with ammonia solution) (65:35, v/v); the flow rate was 0.2 ml/min. The analytical column was kept at 30°C and the effluent was connected to an electrospray ionization MS interface without splitting. Electrospray ionization was performed using nitrogen at 10 l/min flow rate, 40 psi nebulizing pressure, and 350°C drying gas temperature. Capillary voltage was set at 3000 V. Fragment voltage was applied between the capillary outlet and the first skimmer-produced fragment ions by insource collision-induced dissociation by nitrogen. 70 V optimum fragment voltage was selected (varying 50-150 V). The Limit of Determination (LoD) was 5 μg/ml; the Lover Limit of Quantitation (LloQ) was 20 μg/ml; the limit used for data certification was 50 μg/ml. |
| |
| Safety Assessment: Safety was assessed at screening (Visit 1) and at the end of study (Visit 4) by vital signs, a physical examination (general appearance, skin, eyes, ENT, chest/lungs, heart/cardiovascular, muscular-skeletal system, abdomen, kidney, lymph nodes, nervous system, genitourinary and endocrine system) and routine laboratory tests: haematology: erythrosedimentation rate (ESR), red blood cell count (RBC), white blood cells total and differential count (WBC), haemoglobin, haematocrit, platelet count, prothrombin time; clinical chemistry: plasma aspartate aminotransferase (AST), alanine aminotransferase (ALT), gamma-glutamyl transpeptidase (γ-GT), lactate dehydrogenase (LDH), alkaline phosphatase (AP), glucose, creatinine, total serum proteins, sodium and potassium; urinalysis: specific weight, pH, glucose, ketones, haemoglobin, protein, bilirubin, urobilinogen, nitrites. At screening (Visit 1) only virology: hepatitis B antigen (HBsAg), hepatitis C antibody (HCV-Ab), human immunodeficiency virus 1 and 2 antibodies (HIV-1 and -2 Ab), pregnancy test. Adverse events were monitored throughout the study. |
| |
| Statistical analysis: Statistical analyses were performed using the commercially available computer programme Origin (version 7.0). All data are presented as mean ± standard error (SE). Descriptive statistics of the concentration-time data of the two formulation of α-lipoic acid acid were carried out. Pharmacokinetic data are presented as mean ± SEM. To compare the means of continuous parameter one-way ANOVA test was used, while for non-continuous parameters such as T max and half life, a two sided t-test was performed. |
| |
| Results |
| |
| Pharmacokinetic Analysis |
| |
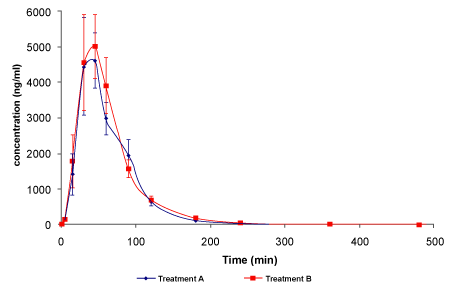
| All the data of the plasma concentration of the two treatments were collected and the pharmacokinetic parameters were calculated assuming the value of 0 for the data under the limit of revelation (50μg/ ml). The mean concentrations of the two drugs in the healthy volunteers were calculated for each sampling point and the concentration curves were built (Figure 1). |
| |
|
|
Figure 1: Concentration/Time Curves of the two formulation of a-lipoic acid. Values are the mean ± Standard Error |
|
| |
| The values of the pharmacokinetics parameter Cmax were 6.66 ± 1.17 μg/ml for treatment A and 7.00 ± 1.32 μg/ml for treatment B with a coefficient of variation percent (CV%) respectively of 74.4% and 80.0%. AUC t was 5.23 ± 7.44 mg/ml*h for treatment A and 5, 77 ± 0, 81 μg/ ml*h for treatment B (CV of 60.3% and 59.9%); AUCinf were 5.65 ± 7.45 μg/ml*h for treatment A and 6.06 ± 0.79 μg/ml*h for treatment B (CV of 55.9% and 55.5%). Tmax parameter were 45.0 ± 4.4 min for the treatment A and 51.8 ± 4.3 for treatment B (CV of 41.2% and 35.4%), while half-life was 46.2 ± 15.1 min for treatment A and 40.2 ± 6.3 for treatment B (CV of 138.8% and 66.4%). |
| |
| Safety evaluation |
| |
| No adverse events were observed during the study. All the subjects examined showed normal values in both screening and final visit. Vital signs, clinical examination and objective laboratory tests were normal at the beginning and the end of the study. |
| |
| No subject left the study due to adverse events or the investigator’s decision. |
| |
| Statistical analysis |
| |
| The statistical analysis of the pharmacokinetics parameter showed no significant difference between the two formulations (Table 4). |
| |
|
|
Table 4: Pharmacokinetical parameters of the two formulation of α-lipoic acid (Data ± SEM). |
|
| |
| Discussion |
| |
| Even though a normal diet contains ALA, food sources fail to provide a pharmacologically active quantity of antioxidant as only small quantities present in food can be absorbed as free ALA [10]. Furthermore, pharmacokinetic analysis of an endogenous substance (as in the case of ALA) is complicated by the interference of basal levels and/or metabolism of the substance. These represent limiting factors in obtaining precise and reliable analyses of plasma levels of lipoic acid or its metabolites, and help to explain the incongruities between published data. Therefore, the analysis of ALA in biological samples has demanded new and complex methodological approaches [11,12]. The analytical procedure used in this study is one of the most reliable methods for identifying ALA plasma levels. In clinical practice, lipoic acid is administered in racemic form (rac-ALA), the pharmacokinetic characteristics of which have already been studied in man after single dose administration [11,13,14,15]. Some studies have examined how food intake and severe renal damage influence the pharmacokinetic parameters of rac-ALA [13,16]. |
| |
| In this study, the Cmax of ALA new formulation was higher and was reached about 15 minutes later than reported in healthy volunteers after a single oral administration of rac-ALA 600 mg [17-20], implying that the galenic formulation influences absorption and absorption time. For example the rate of absorption of lipoic acid is not substantially influenced by the time of gastric emptying, as demonstrated in studies in insulin-dependent diabetics with habitual delayed gastric emptying, in which no important influence on ALA bioavailability was observed [ 14]. A clear relationship between bioavailability and the antioxidative pharmacological effects has not yet been established. The good Cmax indicates that new formulation positively affects absorption and absorption time regardless of the pharmaceutical form. |
| |
| The AUCinf values of new formulation are not high, in accordance with the half-life value, which does not depend on the dose but is correlated to the general conditions of the organs and systems. Taking into consideration the fact that a present is a cross-over study, it seems evident that ALA in softgel capsules and tablets is absorbed more consistently and rapidly and is eliminated efficiently. The AUC value of two treatments is a more reliable measure of ALA bioavailability, since it is directly related to the total amount of non-modified drug which reaches systemic circulation. Careful assay and complete observation of drug elimination were made possible in this study through blood sample collection. |
| |
| Some studies report that the average plasma concentrations at which the therapeutic effects of lipoic acid begin to be seen correspond to Cmax and AUC values equivalent to 4-5 μg/ml and 2.85 μg/ml*h, respectively [12,15,19,21]. Nevertheless, a clear relationship between bioavailability and the antioxidative pharmacological effects has not yet been established. |
| |
| The dose-independent half-life correlated to the general conditions of the organs and systems, further indicating that new formulation is absorbed consistently and rapidly and is eliminated efficiently. |
| |
| Examining the correlation between pharmacokinetic parameters and therapeutic efficacy makes it possible to acquire important information for designing preclinical and clinical studies. The pharmacokinetic characteristics of a compound can significantly limit its clinical use if pharmacologically active concentrations are not reached and/or maintained for the time necessary to evoke therapeutic effects. Since it is not possible to correlate the pharmacokinetics and pharmacodynamics of ALA [21], the therapeutic effects depend preponderantly on the Cmax and AUC values rather than on the time values for reaching maximum concentration (Tmax), elimination halflife (T 1/2) or median retention time (MRT). This opinion is supported by pharmacokinetic studies conducted in patients, which demonstrated that therapeutic response is positively correlated to the Cmax value [22-24]. |
| |
| The antioxidant capacity of ALA depends fundamentally on the induction of glutathione regeneration at the cellular level. The increase of glutathione after ALA administration is probably due to an adaptive cellular response induced by improved employment of the cysteine accumulated in the cell [25]. It has recently been reported that the cellular kinetics of ALA are proportional to the plasma kinetics of the compound [18,20]. Considering the time needed to regenerate cellular glutathione after administration of ALA, new formulation in two different forms could prove to be an able to reach its target quickly at active concentrations and could serve as an excellent point of departure for pursuing further interesting pharmacological and therapeutic developments. The properties of the new formulation ensure rapid release in vivo with high ALA content per unit dose and excellent homogeneity of release. |
| |
| ALA solubility may be increased by vehiculation in an amphiphilic matrix like lecithin, a good emulsifier. After rapid dissolution there is a good solubilization by lecithin: an inexpensive method that is very simple to use. The pharmacokinetic characteristics of tablets and softgel capsules, both delivering new ALA formulation, show the same human pharmacokinetic profile. Rapid release of ALA is achieved through the high speed of solubilization of the soft gelatin and tablets by the action of the superdisaggregating agents. The results should prove useful in treating inflammatory processes and osteoarticular neuropathies, in particular canalicular syndromes, sensory or motor radiculopathies, diabetic polyneuropathies, and peripherical neuropathies of various origins. |
| |
| Acknowledgments |
| |
| The authors thank the Chelab Laboratory for their help in determining α-lipoic acid plasma concentrations and the Medi Service group for their support on data management. |
| |
| |
| References |
| |
- Reed LJ, DeBusk BG, Gunsalus IC, Hornberger CS Jr (1951) Crystalline a-lipoic acid; a catalytic agent associated with pyruvate dehydrogenase. Science 114: 93-94.
- Loffelhardt S, Bonaventura C, Locher M, Borbe HO, Bisswanger H (1995) Interaction of a-lipoic acid enantiomers and homologues with the enzyme components of the mammalian pyruvate dehydrogenase complex. Biochem Pharmacol 50: 637-646.
- Milne JL, Wu X, Borgnia MJ, Lengyel JS, Brooks BR, et al. (2006) Molecular structure of a 9-MDa icosahedral pyruvate dehydrogenase subcomplex containing the E2 and E3 enzymes using cryoelectron microscopy. J Biol Chem 281: 4364-4370.
- Maczurek A, Hager K, Kenklies M, Sharman M, Martins R, et al. (2008) Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer's disease. Adv Drug Deliv Rev 60: 1463-1470.
- Walden HR, Kirby JA, Yeaman SJ, Gray J, Jones DE, et al. (2008) Xenobiotic incorporation into pyruvate dehydrogenase complex can occur via the exogenous lipoylation pathway. Hepatology 48: 1874-1884.
- Schonauer MS, Kastaniotis AJ, Kursu VA, Hiltunen JK, Dieckmann CL (2009) Lipoic acid synthesis and attachment in yeast mitochondria. J Biol Chem 284: 23234-23242.
- Arner ESJ, Nordberg J, Holmgren A (1996) Efficient reduction of lipoamide and lipoic acid by mammalian thioredoxin reductase. Biochem Biophys Res Commun 225: 268-274.
- Bilska A, Wlodek L (2005) Lipoic acid-the drug of the future? Pharmacol Rep 57: 570-577.
- Mignini F, Capacchietti M, Napolioni V, Reggiardo G, Fasani R, et al. (2011) Single dose bioavailability and pharmacokinetic study of a innovative formulation of a-lipoic acid (ALA600) in healthy volunteers. Minerva Med 102: 1-8.
- Packer L (1994) Antioxidant properties of lipoic acid and its therapeutic effects in prevention of diabetes complications and cataracts. Ann NY Acad Sci 738: 257-264.
- Teichert J, Kern J, Tritschler HJ, Ulrich H, Preiss R (1998) Investigations on the Pharmacokinetics of a-lipoic acid in healthy volunteers. Int J Clin Pharmacol Ther 36: 625-628.
- Chen J, Jiang W, Cai J, Tao W, Gao X, Jiang X (2005) Quantification of lipoic acid in plasma by high-performance liquid chromatography-electrospray ionization mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 824: 249-257.
- Gleiter CH, Schug BS, Hermann R, Elze M, Blume HH, et al. (1996) Influence of food intake on the bioavailability of thioctic acid enantiomers (letter). Eur J Clin Pharmacol 50: 513-514.
- Hermann R, Niebch G, Borbe HO, Fieger H, Ruus P, et al. (1996) Enantio selective pharmacokinetics and bioavailability of different racemic a-lipoic acid formulations in healthy volunteers. Eur J Pharm Sci 4: 167-174.
- Breithaupt-Grogler K, Niebch G, Schneider E, Erb K, Hermann R, et al. (1999) Dose-proportionality of oral thioctic acid-coincidence of assessments via pooled plasma and individual data. Eur J Clin Pharmacol Sci 8: 57-65.
- Teichert J, Tuemmers T, Achenbach H, Preiss C, Hermann R, et al. (2005) Pharmacokinetics of a-lipoic acid in subjects with severe kidney damage and end-stage renal disease. J Clin Pharmacol 45: 313-328.
- Teichert J, Hermann R, Ruus P, Preiss R (2003) Plasma kinetics, metabolism, and urinary excretion of a-lipoic acid following oral administration in healthy volunteers. J Clin Pharmacol 43: 1257-1267.
- Mignini F, Streccioni V, Tomassoni D, Traini E, Amenta F (2007) Comparative crossover, randomised, open-label bioequivalence study on the bioequivalence of two formulations of thioctic acid in healthy volunteers. Clin Exp Hypertens 29: 575-86.
- Carlson DA, Smith AR, Fisher SJ, Young KL, Packer L (2007) The plasma pharmacokinetics of R-(+)-lipoic acid administered as sodium R-(+)-lipoate to healthy human subjects. Alern Med Rev 12: 343-351.
- Amenta F, Traini E, Tomassoni D, Mignini F (2008) Pharmacokinetics of different formulations of tioctic (a-lipoic) acid in healthy volunteers. Clin Exp Hypertens 30: 767-775.
- Krone D (2002) The pharmacokinetics and pharmacodinamics of R-(+)-Alpha Lipoic Acid. Johann Wolfgang Goethe University, Frankfurt, am Main, Germany.
- Ydav V, Marracci G, Lovera J, Wooddward W, Bogardus K, et al. (2005) Lipoic acid in multiple sclerosis: a pilot study. Mult Scler 11: 159-165.
- Maczurek A, Hager K, Kenklies M, Sharman M, Martins R, et al. (2008) Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer's disease. Adv Drug Deliv Rev 60: 1463-1470.
- Di Geronimo G, Caccese AF, Caruso L, Soldati A, Passaretti U (2009) Treatment of carpal tunnel syndrome with alpha-lipoic acid. Eur Rev Med Pharmacol Sci 13: 133-139.
- Han D, Handelman G, Marcocci L, Sen CK, Roy S, et al. (1997) Lipoic acid increases de novo synthesis of cellular glutathione by improving cystine utilization. Biofactors 6: 321-338.
|
| |
| |