| Research Article |
Open Access |
|
| Chinmoy Ghosh and Bhaswat Chakraborty* |
| Cadila Pharmaceuticals Limited, 1389-Trasad Road, Dholka-387810, Gujarat, India |
| *Corresponding authors: |
Bhaswat Chakraborty
Research and Development
Cadila Pharmaceuticals Limited
1389-Trasad Road, Dholka-387 810
Gujarat, India
Tel: +91-2714-221481/83/84
Fax: +91-2714-221848
E-mail: drb.chakraborty@cadilapharma.co.in |
|
| Â |
| Received October 14, 2012; Published October 30, 2012 |
| Â |
| Citation: Ghosh C, Chakraborty B (2012) LC-MS Methods for Regulated Bioequivalence Studies: Do we Need to Worry about Matrix Effects? 1:424. doi:10.4172/scientificreports.424 |
| Â |
| Copyright: © 2012 Ghosh C, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| Â |
| Abstract |
| Â |
| Matrix effect is a major problem during any LC-MS/MS analysis. Matrix effects are defined as the effect of co-eluting compounds at the retention time of analyte. If any analytical method is prone to matrix effects then the obtained data is not reliable. It is the primary requirement for any analytical method to be reproducible and reliable. There are no specific methods to overcome the matrix effect related issues. Among many techniques, some of the techniques are: using different sample extraction procedure, using SIL-IS, by switching the ionization polarity, by using different ionization techniques i.e. ESI/APCI, changing the anti-coagulant, different chromatographic conditions, etc. Matrix effects can be eliminated completely/partially by adopting any of the single techniques or in combination of these techniques. According to regulatory requirement, it is mandatory to evaluate the matrix effects during method development and validation. Addressing the matrix effects data during any regulatory submission is must. Researchers face problems during incurred sample reanalysis (ISR), as if the analytical method used for ISR analysis is prone to matrix effects, then there are possibilities that the ISR experiment will fail. Failing the ISR experiment will raise the question about the reliability of the applied method, this failure mostly occur because of presence of matrix effect. So, it is the utmost requirement for the researchers to evaluate, address and determine the matrix effect during any LC-MS/MS analysis. |
| Â |
| Keywords |
| Â |
| Matrix effects; LC-MS/MS; Extraction technique; ESI; APCI; HPLC; UPLC |
| Â |
| Introduction |
| Â |
| Bioequivalence studies are not unknown to the pharmaceutical world. It was started almost more than three decades ago from now. Still then the main objective remain the same i.e. the accurate quantification of the administered drug. But, there was a huge improvement and modification in the analytical techniques used now and then. Initially scientists used HPLC with UV/RI/Fluorescence detector, latter on more advanced techniques like ELISA, RIA came into place. But these techniques also had problems in repeatability, sensitivity and reproducibility. To achieve more sensitivity and reliability of the analytical data scientists started to use GC-MS. In the phase of instrumental advancement, scientists found LC-MS/MS as one of the most sensitive, reliable and fast analytical technique for bioequivalence studies. Modern triple quadrupole instruments allow significantly more productivity than the early instruments. They are easier to operate, rugged, stable and compliant software is readily available for quantitation, providing greater throughput. Though from the experience it is observed that modern LC-MS/MS is prone to some problems and has few limitations. LC flow rate, solvent/reagents, ionization suppression and matrix effects have been shown to be the root cause of many issues for bioanalytical scientists [1]. |
| Â |
| Due to the inherent mass selectivity of tandem mass spectrometric techniques, many interfering sources can be removed by mass filtration [2]. However there are many substances available in the matrix that is not detected in the tandem mass spectrum, but they co-elute with the analyte. These co-eluted matrix components may adversely affect the ionization process and result in significant signal variation. During bio-analysis endogenous components mainly contribute to the interference. As biofluids have heterogeneous variability, the effects of endogenous interferents are not reproducible, which may influence the accuracy and precision of analytical quantification. |
| Â |
| Matrix interferences during LC/MS bio-analysis have been carefully studied. After the study it has been concluded that matrix effects should be evaluated for every newly developed LC/MS methods. Simultaneously regulatory bodies also tighten their screw to the acceptability of the obtained analytical data of bioequivalence studies. Now regulatory bodies are looking for more sensitive, reliable and rugged bioanalytical methods. Recently different regulatory bodies have recommended to perform matrix effects experiment during method validation and incurred sample reanalysis (ISR) during actual study sample analysis. If the ISR experiment fails, then it is recommended to investigate the reason of failure. If the failure is because of the processing error, poor chromatography or instrumental abnormalities then the ISR experiment can be reanalyzed, otherwise it is necessary to redevelop the method. Matrix effects may have a major role to this ISR failure. |
| Â |
| There are lots of reasons of matrix effects during LC-MS/MS bioanalysis. Different strategies are also adopted to overcome or minimize these matrix effects. In this manuscript we will discuss issues related to matrix effects and its way of elimination. Matrix effects related issues will be discussed with examples whenever possible. |
| Â |
| Workshops for guidelines consensus |
| Â |
| Before the first Bioanalytical method validation (BMV) workshop, there was a lack of uniformity in conducting validation of bioanalytical methods and submission of data to regulatory agencies. The bioanalytical validation workshop in 1990 was the first major workshop dedicated to investigating and harmonizing procedures required in method validations [3]. The workshop defined essential parameters for BMV-accuracy, precision, selectivity, sensitivity, reproducibility, limit of quantification, and stability-and addressed “how to†evaluate and determine these parameters. In addition to defining various bioanalytical method validation parameters, the workshop discussed appropriate method validation procedures and defined the standard curve, recovery, and replicate analysis. It was clarified that it is not essential to have 100% recovery, but it is important that the recovery be reproducible. One of the most important outcomes of the first workshop was that it defined “the acceptance criteria for a run.†|
| Â |
| The second workshop focused on discussing the advances in analytical technology that had occurred over the past decade and reconfirmed and updated the principles of BMV [4]. The second workshop discussed the advances in “hyphenated†mass spectrometry and ligand binding assays. Selectivity issues were discussed in detail. Assays must be selective for the analyte. Two types of issues must be considered: (1) interference from substances that are physicochemically similar to analyte (e.g., metabolites, endogenous compounds) and (2) interference from matrix components (also termed “matrix effectsâ€, interference from matrix components that are unrelated to the analyte, such as from homolysis, serum proteins, lipemia, etc) that are unrelated to the analyte. The second workshop also discussed different categories of validation; namely Partial Validation, Cross-Validation, and Full Validation. The workshop reemphasized that it is not necessary to have 100% recovery, but it is important to have reproducible and consistent recovery when using an extraction procedure. The importance of standard curve and quality control acceptance criteria were reemphasized. |
| Â |
| The purpose of the 3rd AAPS/FDA Bioanalytical Workshop was to identify, review, and evaluate the existing practices, white papers, and articles and clarify the FDA Guidance [5]. Bioanalytical methods used to support the drug development process are validated to ensure that they function in the manner in which they are intended. “Incurred†or study samples can vary in their composition when compared with the standards and quality control samples used to validate the method and analyze these samples. During the 3rd American Association of Pharmaceutical Scientists (AAPS)/Food and Drug Administration (FDA) Bioanalytical Workshop, it was suggested that the reproducibility in the analysis of incurred samples be evaluated in addition to the usual pre study validation activities performed. Because it has been observed that, in many occasions bioanalytical methods are unable to reproduce the results in actual study sample reanalysis. The difference in concentration between the original and reanalyzed samples varies from 30-80%, even sometimes more than 200%. These high degrees of variation in concentration data after reanalysis may change the pharmacokinetic/toxicokinetic profile completely. So, to ensure the reliability on the analytical methods and reproducibility of the analytical data, pharmaceutical scientists had recommended performing the incurred sample analysis (ISR). |
| Â |
| Once pharmaceutical scientists had started the analysis of incurred samples, they were facing problem in reproducibility. So, they had started searching the causes behind this irreproducibility, while they identified some matrix components e.g. phospholipids, as a major causes of the irreproducibility, which they had termed as matrix effects. So, during the 3rd American Association of Pharmaceutical Scientists (AAPS)/Food and Drug Administration (FDA) Bioanalytical Workshop, it was suggested that matrix effects will be evaluated during each method development and method validation. |
| Â |
| While developing and validating a Bioanalytical method, it is important to be aware of the bioanalytical risk and the acceptable bioanalytical risk tolerance. Mohammed Jemal had discussed this topic in fourth Bioanalytical workshop, by presenting examples where awareness of certain risks was not very high even 10 years ago [6]. His first example, awareness of the impact of phospholipids on quantitation, was only just becoming a requirement for method development/ validation. It is now necessary to ensure that phospholipids are either separated chromatographically from the analyte of interest, or else that the extraction method is selective enough to remove them from the sample altogether. Another example he provided was the impact of metabolites on quantization. It is possible that a metabolite is isobaric with the parent drug or that it produces the isobaric species via in-source conversion. In such cases, chromatographic separation is essential. It is also possible that back-conversion of the metabolite to the parent compound occurs during the multiple steps involved in sample analysis, including sample handling and extraction. He highly recommended using pooled incurred samples during method development to ensure that metabolites do not impact analyte quantification. In this way, the impact of even previously unknown metabolites can be assessed. It is important to realize that inherent method quality is built during method development. |
| Â |
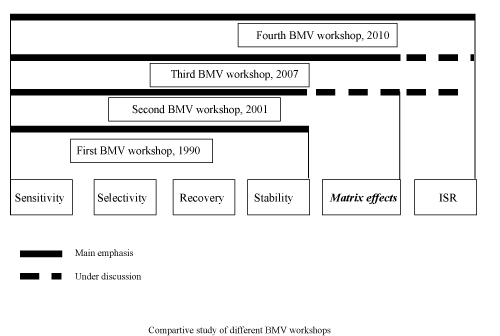
| Many issues were discussed during this workshop. One of the main issues the European Bioanalysis Forum (EBF) presented were matrix effect, study reports, legal basis, ISR, definitions of complete/partial/cross validation, stability and accuracy. For example, EMA specifies that hemolyzed and lipemic lots of matrix must be tested as part of the matrix effect evaluation. The EBF proposed that the test is not always relevant, depending on the molecule and expected use of the method, and therefore should be an option. Figure 1 shows a comparison between discussion agendas in different Bioanalytical workshops. |
| Â |
|
|
Figure 1: Comparison between discussion agendas in different Bioanalytical workshops. |
|
| Â |
| Different causes of matrix effects and how to eliminate |
| Â |
| Different phospholipids, mainly glycerophosphocholines (PC) and lysophosphophatidylcholines (lyso-PC), represent the major class of endogenous phospholipids causing matrix effects [7,8]. Phospholipids are a class of biological compounds having one or more phosphate groups. Molecular structure of phospholipids having two major functional group regions: a polar head group substituent, which includes an ionizable organic phosphate moiety as well as other polar groups of various types, and one or two long chain fatty acid ester groups, which impart considerable hydrophobicity to the molecule [9]. Basically, the highly ionic nature of phospholipids make them responsible for influencing the ionization in ESI-MS [10,11] and desolvation of the LC effluent droplets in ESI-MS analysis [12]. |
| Â |
| Therefore, the removal of these endogenous phospholipids is an extremely important step in the sample preparation process [9]. |
| Â |
| Case-1: Different Sample Processing Techniques |
| Â |
| Analyte can be extracted by different extraction techniques like, protein precipitation, liquid-liquid extraction, solid phase extraction or combination of these. All these techniques will show different extraction recoveries for the same analyte. So it will be a common consideration for the analysts to adopt the extraction technique which have the highest extraction recovery. But this is not always the right decision taking matrix effects into the consideration. Higher degrees of extraction recoveries may be due to the ion enhancement, similarly, ion suppression causing lower extraction recoveries. So, during LC-MS/MS analysis, along with extraction recoveries study of matrix effects is essential before finalizing the sample extraction technique. |
| Â |
| Among all these extraction techniques protein precipitation is more prone to matrix effects, followed by liquid–liquid extraction (LLE) and solid phase extraction, though this is not the thumb rule. In protein precipitation technique, among the commonly used precipitating agents (e.g. methanol, acetonitrile etc.) methanol causing highest degree of matrix effects, as most of the compounds get dissolved into methanol and it is more polar in nature too. It is observed that molecules having higher protein binding causing more matrix effects. |
| Â |
| From several years liquid-liquid extraction technique, which has an edge over other techniques, is preferred for the extraction of smallmolecule drugs and metabolites from plasma samples. Generally, the common practice for LLE is to maintain the plasma pH to 2 units lower than the pKa for acidic molecules and 2 units higher for basic molecules, assuming that the non-ionized analyte species have a better extractability behavior than the ionized species. While this may hold true in general, for some analytes, good extraction efficiency could be achieved under conditions where the analyte is apparently largely ionized. Because of the recent awareness of matrix effects, for development of bioanalytical methods, scientists are studying the fate of phospholipids during the different LLE methods adopted for extraction of drugs and metabolites from plasma samples. Liquidliquid extraction comparatively shows lesser matrix effects. There are many liquid extracting solvents available with wide polarity range. So, sometimes matrix effects can be minimized/eliminated by using solvents of different polarity than the polarity of the molecule of interest, provided sufficient recoveries obtained. Mixture of solvents having different polarities may also be used to overcome this matrix effects problem as well as to increase the overall recoveries. In LLE, scientists were observed that the extraction of the PC and lyso-PC increases with the increase in the polarity of the organic solvents and lyso-PC gets extracted in lesser quantity compared to the PCs. It is also observed that, n-butyl chloride and methyl-tert-butyl ether (MTBE) are the best single component solvents for LLE, considering the minimal extraction of phospholipids mainly lyso phospholipids. Though, phospholipids are more abundant than lyso phospholipids in plasma, but the letter one more significant in LC-MS/MS bioanalysis, as they elute earlier and closer to the commonly used small molecule drugs and their metabolites under the commonly used reversed phase chromatographic conditions. Hence, n- butyl chloride and MTBE is efficient to remove the lyso phospholipids from plasma samples during LLE extraction. |
| Â |
| Another extraction technique which is commonly used is the solid phase extraction (SPE). It is well established that SPE produces the cleanest sample, causing the less matrix effects. Now there are different chemistries are available, like strong cation exchange (SCX) SPE cartridge, strong anion exchange (SAX) SPE cartridge, mixed mode cation exchange (MCX), mixed mode anion exchange (MAX) cartridges etc. By using the SPE technique almost all phospholipids and lyso phospholipids can be eliminated from the sample [13]. |
| Â |
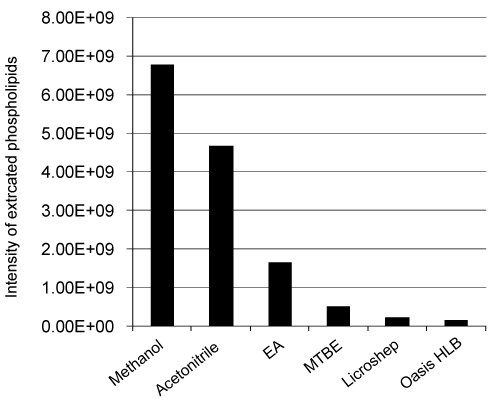
| Nevirapin plasma sample was extracted by using protein precipitation (methanol and acetonitrile as precipitating agent), liquidliquid extraction (Ethyl acetate and MTBE as extraction solvent) and solid phase extraction (SPE cartridges from WATERS Oasis and MERCK Lichrosep). In all these cases, the intensity of extracted phospholipids was presented in figure 2. |
| Â |
|
|
Figure 2: Intensity of phospholipids extracted from plasma samples by different extraction techniques. |
|
| Â |
| One important factor that affects these quantitative performances is ion suppression. Sample matrix, coeluting compounds, and crosstalk can contribute to this effect. Ionization effects can theoretically occur in either the solution phase or the gas phase. Recent experiments involving ESI of biological extracts have shown that the main cause of ion suppression is a change in the spray droplet solution properties caused by the presence of nonvolatile or less volatile solutes [14]. These nonvolatile materials (e.g., salts, ion-pairing agents, endogenous compounds, drugs/metabolites) change the efficiency of droplets by increasing the viscosity and surface tension, thus, reducing solvent evaporation and the ability of the analyte to reach the gas phase [15,16]. These nonvolatile materials can decrease the efficiency of droplet formation through co-precipitation of analyte or preventing droplets from reaching their critical radius required for gas phase ions to be emitted [17], which in turn affects the amount of charged ion in the gas phase that ultimately reaches the detector. |
| Â |
| Case 2: Comparison of Matrix Effects in HPLC vs UPLC |
| Â |
| HPLC is a well known technique to the chromatographic scientists. There is lots of research works is performed for pharmaceutical analysis using HPLC. HPLC is most commonly used liquid chromatographic interface with MS/MS. Scientists use different HPLC columns; mobile phase compositions to reduce/ eliminate the matrix effects, but still sometimes it is not possible to eliminate matrix effects completely. Labeled internal standards, which compensate for (but do not eliminate) matrix effects, were not available for some analytes. The only accurate method for quantification in this manner is standard addition. But the drawback is that this method is labor- intensive and time-consuming. |
| Â |
| But with the advancement of chromatographic sciences, a new technique called ultra performance liquid chromatography (UPLC) is introduced. UPLC technology with lesser particle size (sub-2 μm particles) has demonstrated significant advantages with respect to speed, sensitivity and resolution. Researchers have demonstrated 5-12x increases in speed [18-20], 3-10x improvements in sensitivity [18,19,21] and close to a 2x improvements in resolution [18,20-24]. It was proposed that the added resolution might provide a benefit with respect to matrix effects, through improved separation from endogenous components. |
| Â |
| Chambers E et al. had done the comparison of matrix effects in HPLC and UPLC separations [25]. They evaluated multiple sample preparation techniques, several mobile phase pHs, numerous gradient profiles, and ten different analytes using both HPLC and UPLC systems. Matrix effects were quantitatively determined for each analyte under the various chromatographic conditions and sample preparation methods. The HPLC matrix effects data were paired with their UPLC data run under identical pH, pradient and sample preparation method. Fortysix sets of paired data were then subjected to a paired t-test to determine if there was a statistically significant difference between the two data populations. The paired t-test returned a p (probability) value of 0.0006, objectively indicating that the reduction in matrix effects observed under UPLC conditions in indeed statistically significant, using a value as the threshold value In another experiment, Willy Lambert et al. were performed the comparison of matrix effects in HPLC-MS/MS and UPLC-MS/MS analysis of nine basic pharmaceuticals in surface water [26]. Matrix effects were evaluated on three different surface water samples, collected from three different sources. The results demonstrated that matrix effects were substantially reduced when the UPLC analysis was performed. Matrix interferences coelute less with the analytes than in HPLC analysis, and signal suppression was reduced dramatically. |
| Â |
| Moreover, matrix effects with internal standards were also studied. Normally the internal standard of highest structural similarity to the analyte was chosen which elutes closely with the analyte. Matrix effects were only minor or totally eliminated in UPLC analysis compared with HPLC analysis. The narrower chromatographic peaks, and thus higher peak capacity, generated by the UPLC system increases the resolution of one chromatographic peak from another. For example, an analyte peak would be expected to be better resolved from interference. As a result, the labor-intensive and time consuming standard addition method can be omitted. Even the internal standardization can be used and the overall method is simplified. |
| Â |
| Case 3: Study of Matrix Effects between Different Anticoagulant |
| Â |
| Many methods were adopted to remove matrix effects during LCMS/ MS bioanalysis. Still matrix effects are causing detrimental effect on final analytical data. The decision of which matrix to use for clinical studies can have significant implications. A recent study on matrix effect had observed that anticoagulant present in the plasma samples play an important role over matrix effects. Chin C et al. [27] had examined in details the effect of anticoagulant, lipemia or an interaction between anticoagulant and lipemia, on analyte responses. In their experiment, they analysed Olenzapine and des-methyl Olanzapine along with respective isotopically labeled internal standards. Commercially available lots of K3EDTA and sodium citrate human plasma, and serum from six different donors and sodium heparin human plasma from nine individual donors were normally selected for the experiment. The lipid content of all those matrixes was different. Phosphate buffered saline (PBS) was used as control matrix. The responses of analytes and internal standards from specificity samples prepared using plasma in various lipemic categories were highly variable. This variability reflects extraction and ionization variability combined. High variability and low response occurred more frequently with severely lipemic samples. The overall %CV for each anticoagulant group was more than 27. These results indicate how variable the responses might be from a group of individual subjects with varying degrees of lipid content, the potential variability between individuals with different lipid concentrations or intraindividual variability from sampling at different time points, perhaps after a patient has ingested a meal high in fat. The analyte to internal standard ratios were also inconsistent which indicates that the labeled internal standards were not effectively able to correct the variability in analyte response. The absolute recovery for all of the specificity samples were below 70%. Considerable differences were observed between the recoveries of the analytes and internal standards for some samples, prepared in sodium heparin plasma with high lipemia. This suggested that internal standards had not effectively mimicked the behavior of the analytes either during extraction. Lipemia may not have been solely responsible for the internal standard tracking problem because difference occurred with plasma having high as well as low lipemic categories. In an attempt to elucidate the cause of the lower recovery for some samples, internal standard recovery was separated into absolute extraction and instrumental recovery. Absolute extraction recovery of the internal standards was calculated by comparing the absolute areas from blanks and samples fortified with internal standard to blanks fortified internal standard post extraction. Extraction recovery ranged from 40-90% for most of the samples, though some samples prepared in sodium heparin, sodium citrate, K3EDTA and serum with high lipemia showed recovery of 20- 30% for both internal standards. This was evidence that lipemia was not the only factor effecting the extraction recovery. Anticoagulant and lipemia were determined to significantly affect instrumental matrix and overall extraction matrix effects. |
| Â |
| In another experiment, Smeraglia J et al. [28] was observed that plasma samples having lithium heparin as anticoagulant showed more matrix effect in comparison with plasma samples having EDTA as anticoagulant. Drug spiked into lithium heparinised and EDTA plasma gave peak area ratios which indicated significant differences. The %CV was 24.5 and 8.11 respectively. The result indicates that the primary cause of matrix related irreproducibility was related to the use of lithium heparin as anticoagulant. |
| Â |
| In general, these results suggest that matrix effects, due to matrix components such as anticoagulant and lipids, affect the performance of an assay significantly and should be evaluated during a validation or prior to the analysis of patient samples. |
| Â |
| Case 4: Comparison Between Different Bio-Fluids |
| Â |
| The presence of matrix effects also proved to be dependent on the bio-fluid analyzed. Matrix components, characteristic to each biofluid, interfered at different times and a varying extent throughout the chromatographic analysis. The major interferences in urine proved to be hydrophilic residual components, most likely inorganic salts. Oral fluid had more interferences than urine, mainly in ESI, and residual matrix components were of a hydrophilic and hydrophobic nature, including proteins, amino acids, and especially mucin. Finally, for plasma, residual matrix components also had a wide polarity range, but their concentrations were apparently higher than those of oral fluid. So matrix effect was dependent on ionization type, sample preparation, and bio-fluid type [11]. |
| Â |
| Case 5: Role of Isotopically Labeled Internal Standard on Matrix Effects |
| Â |
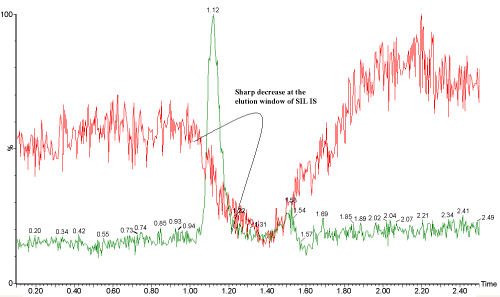
| A stable isotopically labeled (SIL) analogue is believed to be the most appropriate internal standard in quantitative bioanalytical LC-MS/MS assay. It is assumed that a SIL internal standard always compensates for variability in chemical derivatization, sample extraction and LC-MS/MS analysis due to its nearly identical chemical and physical properties to the unlabeled analyte. In theory, since the SIL is almost identical in structure to and co-elute with the analyte, the degree of ionization suppression or enhancement caused by the coeluting matrix components should be the same for the IS and analyte. Therefore, while the absolute response may be affected, the analyte to IS peak area ratio should be unaffected and the bioanalytical method should be accurate, precise and rugged. But this is not always true that matrix effects may be entirely compensated if a SIL internal standard is used. The deuterium isotope effect may cause the partial resolution of analyte and its deuterated IS in reversed phase LC. If a large and sharp matrix suppression peak elutes at around the retention time of analyte and IS, the slight difference of retention time may cause differential matrix effects and affect the accuracy and precision of the quantitative bioanalytical analysis (Figure 3) [29]. |
| |
| Â |
|
|
Figure 3: SIL-IS elutes at the slope of ion suppression. |
|
| Â |
| Case 6: Role of Organic Solvents on Matrix Effects |
| Â |
| Matrix effects can profoundly reduce the performance of electrospray ionization mass spectrometry. It was observed that organic solvents used in mobile phases and extract preparation of biological samples may be associated with the ion suppression, affecting adduct formation and assay sensitivity [30]. Preliminary observations indicated that the methanol used in the mobile phase could be a source of differential ionization or ion suppression. |
| Â |
| From the above literature review it was observed that, the impact of matrix effects on the accuracy, precision and robustness of bioanalytical methods is of growing concern in the pharmaceutical industry. Residual matrix components, endogenous phospholipids in particular, are a significant source of imprecision in quantitative analyses commonly conducted by LC- MS/MS. Matrix effects result from co-eluting matrix components that affect the ionization of the target analyte, resulting either ion suppression, or, in some cases, ion enhancement. Matrix effects can be highly variable and can be difficult to control or predict. They are caused by numerous factors, including, but not limited to endogenous phospholipids, dosing media, formulation agents and mobile phase modifiers. Matrix effects can be compounded by coeluting metabolites, impurities or degradation products. Furthermore, matrix effects are analyte specific. Current FDA guidance documents now require that these effects be evaluated as a part of quantitative LCMS/ MS method development, validation and routine use [31-34]. |
| Â |
| Since, matrix effects is analyte specific and there are no fixed procedure to overcome the matrix effects related issues, so still there are lots of scopes for research on matrix effects. There may be many other novel causes of matrix effects which we need to find out. Moreover, there is no standard and systematic procedure towards identification and quantification of matrix effects in biological matrix during LC-MS/MS bioanalysis. |
| Â |
| Case 7: Ionization Polarity Causing Matrix Effects |
| Â |
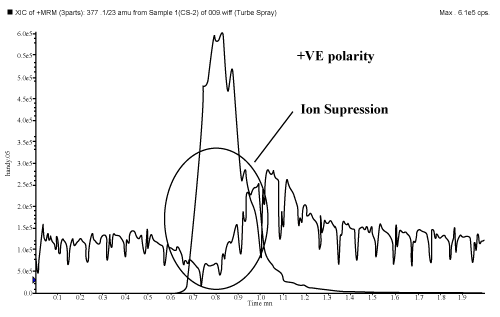
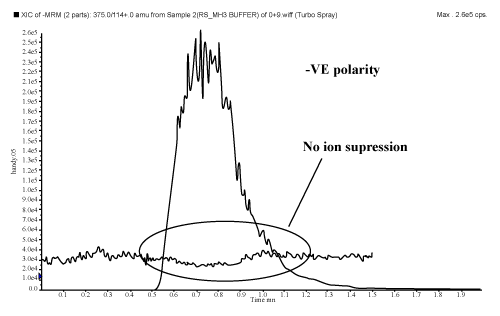
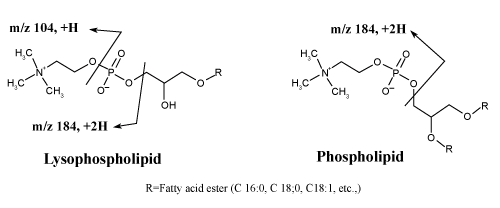
| Playing with ionization polarity is one of the techniques to overcome/ minimize the matrix effects. All molecules either produce positive or negative ions inside the ion source to get analyzed and detected. Some molecules get ionized in both the polarity i.e. zwitter ions. During LC-MS/MS analysis it is observed that most of the interfering compounds e.g. phospholipids are get detected in positive polarity. So, LC-MS/MS analysis in positive polarity is more susceptible to matrix effects. It is recommended to check the matrix effects prior initiation of method validation and during method development, if matrix effect exists and the intended molecule can ionized in both the polarity, then trials should be carried out in opposite polarity too, provided the responses are sufficient to detect the LLOQ level. Chinmoy Ghosh et al. had performed an experiment where they had shown that by changing the polarity matrix effects can be eliminated/ minimized by using enalapril molecule. Enalapril showed more matrix effects in positive ionization mode (Figure 4) in comparison with negative mode (Figure 5), because most of the phospholipids are ionized in positive polarity due to the presence of tertiary amine, which easily ionizes in positive mode (Figure 6). These phospholipids affect the efficiency of gas phase ion formation from the liquid phase. |
| Â |
|
|
Figure 4: Enalapril shows ion suppression in positive polarity. |
|
| Â |
|
|
Figure 5: Enalapril shows no matrix effects in negative polarity. |
|
| Â |
|
|
Figure 6: Fragmentation pathways of phospholipids. |
|
| Â |
| Case 8: Study of Matrix Effects in Different Ionization Techniques (ESI vs APCI) |
| Â |
| Many different mechanisms of ion suppression have been proposed. Different ionization techniques were also responsible for matrix effects. Two most popular ionization techniques are electrospray ionization (ESI) and atmospheric pressure chemical ionization (APCI). |
| Â |
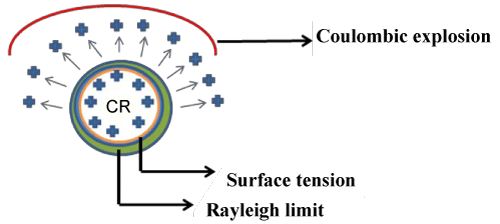
| The ionization mechanism is totally different for ESI and APCI. In ESI, once air born, the liquid droplets’ structural integrity becomes dependent upon the struggle of surface tension with the electrostatic repulsion that results from the solvated ions. Upto a point, known as Rayligh limit, surface tension will hold the repulsive forces in check and prevent droplet fragmentation. Due to evaporation, however, continuous shrinkage in the droplet size gradually brings the charges closer together, increasing repulsion proportionately. Eventually, the Rayligh limit is overcome and the droplet undergoes columbic explosion, splitting into progeny droplets in which the process is reset (Figure 7). |
| Â |
|
|
Figure 7: Ionization mechanism in ESI-MS. |
|
| Â |
| Electrospray is a sensitive detection method for polar molecules. But, at high concentrations (>10-5 M), the approximate linearity of the ESI response often lost [30]. The loss of response may be due to a limited amount of excess charge available on ESI droplets or because of the saturation of the ESI droplets with analyte at their surfaces at high analyte concentrations, causing inhibition of ejection of ions trapped inside the droplets. Regardless of the mechanism for saturation, in multi component samples of high concentrations, competition for either space or charge most likely is occurring and, in turn, suppression of signal is observed. Both the characteristics and concentration of an analyte determine the efficiency of its ionization. The characteristics that decide whether a compound will out-compete others for the limited charge or space on the surface of the droplet include its surface activity and its basicity. Since, biological matrices contain large amounts of endogenous compounds with potentially very high basicities and surface activities, but limit concentration of 10-5 M of ions is reached quickly, and ion suppression occurs with such samples [35]. There is only so much charge that a droplet can hold and all the species are competing for that charge. So if a large concentration of another species co-elutes with one of the drug metabolites, the competition will depress ionization of the target molecule, causing ion suppression. |
| Â |
| APCI frequently gives rise to less ion suppression than ESI, because of different ionization mechanisms [36,37]. As a result, the degree of ion suppression often is different for ESI and APCI, in the presence of other coeluted compounds. Unlike ESI, there is no competition between analytes to enter the gas phase, because neutral analytes are transferred into the gas phase by vaporizing the liquid in a heated gas stream. Moreover, in APCI ion suppression is directly related to charge saturation, because the maximum number of ions formed by gas-phase ionization is much higher, as reagent ions are redundantly formed [37]. Nonetheless, APCI does experience ion suppression, which has been explained by considering the effect of sample composition on the efficiency of charge transfer from the corona discharge needle [38]. In addition, because there is very little chance for analytes to pass through the vaporization region and remain in solution, another mechanism of ion suppression in APCI is solid formation, either as pure analyte or as a solid coprecipitate with other nonvolatile sample components [39]. |
| Â |
| Conclusion |
| Â |
| Although LC-MS/MS is one of the most sensitive and selective analytical and detection technique, but matrix effects can play a detrimental role towards the final outcome of the analysis. So, it is the utmost requirement for the scientists, who are working on LC-MS/ MS, to address this issue every time they developed any new methods. Though there are no universal solutions available, but different strategies are needed to be adopted for different molecules or plasma lots to reduce the matrix effects. Different techniques are available to overcome/ eliminate the matrix effects, but it requires careful application of these techniques. During the audit by different regulatory bodies, they are focusing more on experiments related to matrix effects, so, it is the time for researchers to take this issue seriously. |
| Â |
| |
| References |
| Â |
- Bennett P (2011) Future developments for triple quadrupole mass spectrometers: not just hardware. Bioanalysis 3: 709-711.
- Du L, White RL (2008) Reducing glycerophosphocholine lipid matrix interference effects in biological fluid assays by using high-turbulence liquid chromatography. Rapid Commun Mass Spectrom 22: 3362-3370.
- Shah VP, Midha KK, Dighe S, McGilveray IJ, Skelly JP, et al. (1991) Analytical methods validation: bioavailability, bioequivalence and pharmacokinetic studies. Conference report. Eur J Drug Metab Pharmacokinet 16: 249-255.
- Guidance for Industry; Bioanalytical Method Validation; USFDA; CDER; 2001.
- Viswanathan CT, Bansal S, Booth B, DeStefano AJ, Rose MJ, et al. (2007) Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays. Pharm Res 24: 1962-1973.
- Savoie N, Garofolo F, van Amsterdam P, Bansal S, Beaver C, et al. (2010) White Paper on Recent Issues in Regulated Bioanalysis & Global Harmonization of Bioanalytical Guidance. Bioanalysis 2: 1945-1960.
- Ismaiel OA, Halquist MS, Elmamly MY, Shalaby A, Karnes HT (2007) Monitoring phospholipids for assessment of matrix effects in a liquid chromatography-tandem mass spectrometry method for hydrocodone and pseudoephedrine in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 859: 84-93.
- Little JL, Wempe MF, Buchanan CM (2006) Liquid chromatography-mass spectrometry/mass spectrometry method development for drug metabolism studies: Examining lipid matrix ionization effects in plasma. J Chromatogr B Analyt Technol Biomed Life Sci 833: 219-230.
- Pucci V, Di Palma S, Alfieri A, Bonelli F, Monteagudo E (2009) A novel strategy for reducing phospholipids-based matrix effect in LC-ESI-MS bioanalysis by means of HybridSPE. J Pharm Biomed Anal 50: 867-871.
- Mei H, in: Korfmacher W (Ed.), Using Mass Spectrometry for Drug Metabolism Studies, CRC Press, Boca Raton, FL, 2005, 103-150.
- Shen JX, Motyka RJ, Roach JP, Hayes RN (2005) Minimization of ion suppression in LC-MS/MS analysis through the application of strong cation exchange solid-phase extraction (SCX-SPE). J Pharm Biomed Anal 37: 359-367.
- King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T (2000) Mechanistic investigation of ionization suppression in electrospray ionization. J Am Soc Mass Spectrom 11: 942-950.
- Ghosh C, Gaur S, Shinde CP, Chakraborty B (2011) Asystematic approach to overcome the matrix effect during LC-ESI-MS/MS analysis by different sample extraction techniques; J Bioequiv Availab 3: 122-127.
- King R, Bonfiglio R, Fernandez-Metzler C, Miller-Stein C, Olah T (2000) Mechanistic investigation of ionization suppression in electrospray ionization. J Am Soc Mass Spectrom 11: 942-950.
- Mallet CR, Lu Z, Mazzeo JR (2004) A study of ion suppression effects in electrospray ionization from mobile phase additives and solid-phase extracts. Rapid Commun Mass Spectrom 18: 49-58.
- Antignac JP, Wasch KD, Monteau F, Brabander HD, Andre F, et al. (2005) The ion suppression phenomenon in liquid chromatography-mass spectrometry and its consequences in the field of residue analysis; Analytical Chemistry Acta 529: 129–136.
- Stahnke H, Alder L, Reemtsma T (2008) No More Trouble with Matrix Effects? Federal Institute for Risk Management; 45th Florida Pesticide Residue Workshop.
- Churchwell MI, Twaddle NC, Meeker LR, Doerge DR (2005) Improving LC-MS sensitivity through increases in chromatographic performance: comparisons of UPLC-ES/MS/MS to HPLC-ES/MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci 825: 134-143.
- Wilson ID, Nicholson JK, Castro-Perez J, Granger JH, Johnson KA, et al. (2005) High resolution "ultra performance" liquid chromatography coupled to oa-TOF mass spectrometry as a tool for differential metabolic pathway profiling in functional genomic studies. J Proteome Res 4: 591-598.
- Lurie IS (2005) High-performance liquid chromatography of seized drugs at elevated pressure with 1.7 microm hybrid C18 stationary phase columns. J Chromatogr A 1100: 168-175.
- Plumb R, Castro-Perez J, Granger J, Beattie I, Joncour K, et al. (2004) Ultra-performance liquid chromatography coupled to quadrupole-orthogonal time-of-flight mass spectrometry. Rapid Commun Mass Spectrom.18: 2331-2337.
- Shen JX, Wang H, Tadros S, Hayes RN (2006) Orthogonal extraction/chromatography and UPLC, two powerful new techniques for bioanalytical quantitation of desloratadine and 3-hydroxydesloratadine at 25 pg/mL. J Pharm Biomed Anal 40: 689-706.
- Leandro CC, Hancock P, Fussell RJ, Keely BJ (2006) Comparison of ultra-performance liquid chromatography and high-performance liquid chromatography for the determination of priority pesticides in baby foods by tandem quadrupole mass spectrometry. J Chromatogr A 1103: 94-101.
- Novakova L, Matysova L, Solich P (2006) Advantages of application of UPLC in pharmaceutical analysis. Talanta 68: 908-918.
- Chambers E, Wagrowski-Diehl DM, Lu Z, Mazzeo JR (2007) Systematic and comprehensive strategy for reducing matrix effects in LC/MS/MS analyses. J Chromatogr B Analyt Technol Biomed Life Sci 852: 22-34.
- Van De Steene JC, Lambert WE (2008) Comparison of matrix effects in HPLC-MS/MS and UPLC-MS/MS analysis of nine basic pharmaceuticals in surface waters. J Am Soc Mass Spectrom 19: 713-718.
- Chin C, Zhang ZP, Karnes HT (2004) A study of matrix effects on an LC/MS/MS assay for olanzapine and desmethyl olanzapine. J Pharm Biomed Anal 35: 1149-1167.
- Smeraglia J, Baldrey SF, Watson D (2002) Matrix Effects and Selectivity Issues in LC-MS-MS. Chromatographia 55: S95-S99.
- Wang S, Cyronak M, Yang E (2007) Does a stable isotopically labeled internal standard always correct analyte response? A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. J Pharm Biomed Anal 43: 701-707.
- Annesley TM (2007) Methanol-associated matrix effects in electrospray ionization tandem mass spectrometry. Clin Chem 53: 1827-1834.
- Bennett P, Liang H Overcoming Matrix Effects Resulting from Biological Phospholipids Through Selective Extractions in Quantitative LC/MS/MS; Presented at the 2004 ASMS Conference, Nashville, Tennesee; Tandem Capabilities Publications, available at https://www.tandemlabs.com/documents/PatrickASMSPaper.pdf.
- Meng M, Bennett P, Tandem Capabilities Publications, available at https://www.tandemlabs.com/documents/MinASMSPaper.pdf.
- Larger PJ, Breda M, Fraier D, Hughes H, James CA (2005) Ion-suppression effects in liquid chromatography-tandem mass spectrometry due to a formulation agent, a case study in drug discovery bioanalysis. J Pharm Biomed Anal 39: 206-216.
- Ahnoff M, Hagelin H (2004) Matrix Effects in Electrospray Ionization: Characterization of Plasma Phospholipids as Suppressors/Enhancers of Ionization Efficiency; Poster session handout at the 52nd American Society for Mass Spectrometry; ASMS Conference on Mass Spectrometry and Allied Topics, Nashville, TN.
- Kebarle P, Tang L (1993) Dependence of ion intensity in electrospray mass spectrometry on the concentration of the analytes in the electro sprayed solution. Anaytical Chemistry 65: 972A–986A.
- Iribarne JV, Thomson BA (1976) On the Evaporation of Small Ions from Charged Droplets. Journal of Chemical Physics 64: 2287-2294.
- Thomson BA, Iribarne JV (1979) Field induced ion evaporation from liquid surfaces at atmospheric pressure. Journal of Chemical Physics 71: 4451-4463.
- Blades AT, Ikonomou MG, Kebarle P (1991) Mechanism of electrospray mass spectrometry Electrospray as an electrolysis cell; Analytical Chemistry 63: 2109-2114.
- Van Berkel GJ (1997) Electrospray Ionization Mass Spectrometry, Fundamentals, Instrumentation & Applications; Cole, R. B., Ed.; Wiley-Interscience: NewYork, Chapter 2.
|
| Â |
| Â |