| Research Article |
Open Access |
|
Yuqiao Sheng1, Jie Zhao2, Yidong Li1, Shuhong Liang1, Suke Sun1, Feng Zhang1,
Li Yang1, Wenzhe Lv3, Lisa E Diamond3, Charlie Xu1 and Shengjun Zhang1,3* |
| 1Department of Clinical Experimental Research Center, The Frist Affiliated Hospital of Zhengzhou University, Institute of Clinical Medicine Henan Province, Zhengzhou, Henan, China |
| 2Department of Clinical Pharmacy, The Frist Affiliated Hospital of Zhengzhou University, Institute of Clinical Medicine Henan Province, Zhengzhou, Henan, China |
| 3Frontage Laboratories, Inc., Exton, PA, USA |
| *Corresponding author: |
Shengjun Zhang
Department of Clinical Experimental Research Center
The Frist Affiliated Hospital of Zhengzhou University
Institute of Clinical Medicine Henan Province
Zhengzhou, Henan, China
E-mail: szhang@frontagelab.com |
|
| Â |
| Received December 14, 2010; Published October 26, 2012 |
| Â |
| Citation: Sheng Y, Zhao J, Li Y, Liang S, Sun S, et al. (2012) Pharmacokinetics and Bioequivalence of Two Formulations of Letrozole 2.5 mg Tablets: A Randomized, Open-Label, Single-Dose, Two-period, Parallel Study in Healthy Chinese Subjects. 1:435. doi:10.4172/scientificreports.435 |
| Â |
| Copyright: © 2012 Sheng Y, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| Â |
| Abstract |
| Â |
| Objective: To evaluate the pharmacokinetics and bioequivalence of a generic (test) and branded (reference) formulation of letrozole 2.5 mg tablets in healthy Chinese volunteers to meet the criteria for marketing the test drug in China. |
| Â |
| Method: The current study was a randomized, open-label, single-dose, one-period, parallel test under fasted conditions in 26 healthy Chinese female and male volunteers. Blood was sampled at baseline (pre-dose) and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 24, 48, and 72 hours post-dose from subjects randomly had reference or test drugs, respectively. Vital signs including temperature, blood pressure and heart rate were measured before and after dosing. Adverse events were recorded throughout the study. Plasma concentrations of letrozole were determined using a validated LC/MS/MS method. The primary pharmacokinetic parameters, Cmax, Tmax, AUC0-t, AUC0-inf were calculated for comparison of bioequivalence between the test (T) and reference (R) medications. |
| Â |
| Results: All 26 subjects (19 female, 7 male; mean age, 24 years [range, 21-29 years]; weight, 57.5 kg [range, 49.0-67.5 kg]) completed the study and data from them were included for comparison followed. The geometric mean Cmax for the test and reference formulations was 40.124 and 39.608 ng/mL, respectively. The mean AUC0-t was 1,437.933 and 1,532.048 ng•h/mL and the mean AUC0-inf was 2,566.558 and 2,953.276 ng•h/mL. The geometric mean ratios (test:reference) for Cmax and AUClast were 1.01 and 0.93, respectively. And The 90% confidence intervals (CIs) of the ratio of AUClast and Cmax were ranging from 84.97% to 102.31% and 91.97% to 111.30%, respectively. |
| Â |
| Conclusion: Our study on healthy fasted volunteers shows the test letrozole formulation of 2.5 mg tablets and reference formulation are bioequivalent. |
| Â |
| Keywords |
| Â |
| Letrozole; Bioequivalence; Pharmacokinetics; LC/MS/ MS |
| Â |
| Introduction |
| Â |
| Breast cancer is the leading type of cancer among women worldwide and accounts for nearly one in four cases of cancer among women currently [1]. In addition, the nonquantifiable effects of breast cancer in terms of qualities of life and concerns for tumor recurrence and death should not be underestimated [2]. Estrogen signaling is thought to be of primary importance in the proliferation and progression of breast cancer. Recently the aromatase inhibitors, which induce estrogen decline, have proved to be effective and are used for therapy for breast cancer [3]. Letrozole, 4,4'-(1H-1,2,4-Triazol- 1-ylmethylene)-dibenzonitrile, is a triazole which could bind to the haem group of aromatase reversibly [4], and is used for the adjuvant treatment of postmenopausal women with hormone receptor positive early breast cancer, for first-line treatment of postmenopausal women with hormone receptor positive or hormone receptor unknown locally advanced or metastatic breast cancer and for the treatment of advanced breast cancer in postmenopausal women with disease progression following antiestrogen therapy [5]. What’s more, male breast cancer accounts for 1% of all breast cancer cases and 1% of all cancer cases in men [6]. But the incidence has increased by 26% over the last 25 years. At the time of treatment of female patients, Letrozole could be used for therapy for male breast cancer [7]. |
| Â |
| Letrozole is an odorless white to yellowish crystalline powder, which is not soluble in water but in dichloromethane and ethanol. As an FDA-approved third-generation aromatase inhibitor, letrozole is more potent, better tolerated and more selective. A new generic formulation of letrozole 2.5 mg tablets (test, [Jiangsu Hengrui Medicine Co., Ltd., Batch number 09112856]) has been developed. Here healthy Chinese male and female volunteers were involved in comparison of the test formulation to the branded formulation (reference, Femara® 2.5 mg tablets [Letrozole, batch number F4240]). We aimed to demonstrate the bioequivalence between the reference and test formulations of letrozole 2.5 mg oral tablets in healthy Chinese adult subjects under fasted conditions. In this paper we presented the pharmacokinetic data from studies above, and those data were important for approval of the test oral letrozole marketing. |
| Â |
| Methods |
| Â |
| Study design and procedures |
| Â |
| This was a single-centre, randomized, open-label, single-dose, one-period, parallel study to assess the bioequivalence (BE) of reference (R) and test (T) formulations of letrozole 2.5 mg oral tablets in healthy Chinese subjects. The study was performed in accordance with the ethical standards for studies in humans of the Declaration of Helsinki and its amendments [8], the International Conference on Harmonisation Guideline for Good Clinical Practice (ICH) [9], and the Guideline for Good Clinical Practice recommended by the State Food and Drug Administration (SFDA) of China [10]. Randomly subjects were divided into 2 groups and each group accepted one of the treatments (T or R) according to a randomization schedule prepared prior to the start of the study. The subjects were under medical supervision at the monitored facility throughout the study. Before drug administration subjects were admitted into the research center and fasted for 10 hours. On day 1, following the overnight fasting, subjects received a single oral dose of the reference or test formulation of letrozole 2.5 mg tablets with approximately 240 mL water. The subjects were discharged from the research facility after 24 hours and returned to the facility on day 3 and 4 for blood sample collection. For determination of letrozole plasma concentrations and pharmacokinetic (PK) analysis, blood samples were obtained at baseline (pre-dose) and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 24, 48, and 72 hours post-dose. This is an acceptable design for a bioequivalence study in accordance with the relevant Food and Drug Administration (FDA) guidelines [11]. |
| Â |
| The protocol for this study was reviewed and approved by the First Affiliated Hospital of Zhengzhou University Institutional Review Board (Zhengzhou, Henan, China), and the study was conducted at the First Affiliated Hospital of Zhengzhou University Frontage China Clinical Research Center, in Zhengzhou between January 5, 2010 and February 6, 2010. All subjects submitted written informed consents before the study procedure was performed. The nature and risks of this study had been fully explained to each subject, and their rights were informed, including the right to withdraw from the study freely, before the study started. |
| Â |
| Subjects |
| Â |
| In this study healthy Chinese men and nonpregnant women between 18 and 45 years of age and within 15% of ideal weight based on height and body frame were eligible. Subjects were excluded if they were current smokers or users of any tobacco products, had a history of hypersensitivity to letrozole or any other component of the letrozole tablets, had used an investigational drug or product, or participated in a drug research study within a period of 30 days prior to receiving study drug, used of any prescription drug therapy within 14 days prior to receiving study drug, donated blood (1 pint or more) within 30 days or plasma within 7 days prior to receiving study drug, had a recent history of alcoholism within 2 years or use of alcohol within 24 hours prior to receiving the dose of study drug, and had any clinically significant abnormality based on medical history, physical examination and laboratory analysis. |
| Â |
| Sample collection and processing |
| Â |
| As mentioned above, subjects arrived at the research facility and fasted overnight. The seated blood pressures and pulse rates of all subjects were measured before having drugs (within 60 minutes pre-dose) and at 4, 8, 12, 24, 48, and 72 hours post-dose. Blood were sampled at baseline and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 24, 48, and 72 hours post-dose. Besides vital signs (temperature, blood pressure, and heart rate) were assessed when the blood samples of 48-72 hour post-dose were collected. The blood collected was used for clinical laboratory tests (chemistry, hematology). |
| Â |
| Blood samples were collected into chilled blood collection tubes containing K2EDTA solution, and were immediately centrifuged at 2,000 g for 10 min at 4ºC. Plasma was separated and stored at -70ºC until shipped on dry ice to Frontage Laboratories (Shanghai), Inc. (Shanghai, China) for LC/MS/MS analysis. The whole procedure from blood collecting to plasma storage was carried out for less than 60 min. |
| Â |
| Tolerability |
| Â |
| To assess drugs’ tolerability and safety, adverse experiences (AE) were monitored, as well as medical history, medication history, physical examination, vital sign evaluation (sitting blood pressure, pulse rate, height, weight and temperature), resting 12-lead electrocardiogram (ECG) and clinical laboratory tests (chemistry, haematology, urinalysis, hepatitis B & C diagnostic profile and urine pregnancy screen) were evaluated within 21 days prior to participating in study. |
| Â |
| Any AE (clinical sign, symptom, or disease) temporally associated with the use of this investigational agent, whether or not considered to be related to the investigational product, were all documented. All AEs occurred during this study, including during the washout interval, were documented. |
| Â |
| Determination of plasma letrozole |
| Â |
| Plasma samples were extracted using a liquid-liquid extraction technique. Plasma (200 μL) was mixed with 20 μL of diluents of methanol and water (50:50, v/v), 20 μL of internal standard spiking solution (letrozole-d4, 20 ng/ml), 200 μL of 0.1 M NaOH solution and 2 mL of methyl t-butyl ether. The samples were then vortexed for 1 min at 2,500 rpm. Following vortex, samples were centrifuged at 3,500 rpm for 5 min and upper organic layers were transferred to clean test tubes and evaporated to dryness at 40°C under nitrogen flow. Next samples were reconstituted with 200 μL of reconstitution solution containing formic acid, CAN and water (0.1:40:60, v/v/v) and a 10-μL sample was injected to the LC/MS/MS system (Sciex API 4000 with Shimadzu LC pump and autosampler) with Column (Phenomenex, Synergi Polar-RP, 80A, 50×3.0 mm, 4 micron) for determination of letrozole concentration. Data acquisition and processing were powered by the Analyst 1.4.2 software package (Applied Biosystems, Foster City, California). |
| Â |
| Pharmacokinetic and statistical methods |
| Â |
| Letrozole plasma concentrations were summarized for each sampling time using descriptive statistics (mean, standard deviation, coefficient of variation, median, minimum and maximum values). Pharmacokinetic parameters (AUC0-t, AUC0-inf, Cmax, tmax, and t½) were generated based upon scheduled blood collection time using noncompartmental methods by WinNonlin (Version 5.0.1, Pharsight Corporation, Mountain View California, USA). PK parameters were summarized for 2 drugs. In addition to the descriptive statistics listed above, geometric means were reported for the pivotal PK endpoints (AUC0-t, AUC0-inf, and Cmax). |
| Â |
| Bioequivalence of the test and reference formulations was determined based on AUC0-t, AUC0-inf and Cmax of plasma letrozole. The bioequivalence was considered if the 90% confidence intervals (CIs) on the ratios of test to reference formulations were within a range of 80-125%. Log-transformed PK parameters AUC0-t, AUC0-inf, and Cmaxwere analyzed using analysis of variance (ANOVA) model including terms for sequence, formulation, and period as fixed effects, and subject nested within sequence as a random effect. Sequence was tested using subject nested within sequence as the error term. A CI on the ratio of untransformed PK parameters was derived through reverse transformation of the 90% CI for the difference in the log scale to the 90% CI for the ratio in the original scale. Plots of mean concentration levels of plasma letrozole versus time were generated for each treatment group. Individual concentration versus time graphs was also included in this study. |
| Â |
| Results |
| Â |
| Demographic data |
| Â |
| Twenty-six Chinese subjects including 19 female and 7 male volunteers were enrolled in this study, received one dose of study medication, and completed the study. The demographic characteristics of the study population were summarized in Table 1. The average BMI was 20.8. Data from all 26 subjects were included in pharmacokinetic analyses and the evaluation of safety. |
| Â |
|
|
Table 1: Subject demographics (N=26). |
|
| Â |
| Pharmacokinetic analysis |
| Â |
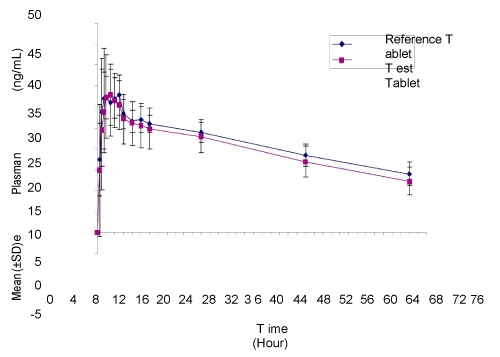
| Linear and semi-log plots of mean letrozole concentration-time profiles after administration of a single 2.5-mg oral dose of test or reference formulations to 13 subjects respectively were presented in Figure 1. Pharmacokinetic analysis of the primary PK parameters (Cmax, Tmax, t1/2, AUC0-t and AUC0-inf) for the test and reference formulations was evaluated with a non-compartmental model using PK features of Wason LIMS (Version 7.3) and presented in Table 2. The mean (± standard deviation [SD]) Cmax values of test and reference formulations were 40.124 (± 5.794) and 39.608 (± 5.330) ng/mL, respectively. The mean (± SD) Tma values of test and reference formulations were 2.167 (± 1.354) and 2.538 (± 1.478) hours. Results for the extent of absorption, as determined from mean (± SD) AUC0-t and AUC0-inf values, were 1,437.933 (± 237.860) and 2,566.558 (± 851.264) ng•hr/mL for test formulation, and 1,532.048 (± 167.114) and 2,953.276 (± 816.906) ng•hr/mL for reference formulation. The mean (± SD) t1/2 of test and reference formulations were 58.758 (± 24.457) and 66.758 (± 23.015) hours. |
| Â |
|
|
Figure 1: Mean (±SD) letrozole plasma concentration vs. time profile (linear plot) after administration of a single 2.5-mg oral dose of test (Jiangsu Hengrui Medicine Co., Ltd., batch number 09112856) and reference (Femara® letrozole 2.5 mg tablets, Novartis Pharmaceuticals Corporation East Hanover, batch number F4240) formulations in healthy Chinese female and male subjects (N=13). |
|
| Â |
|
|
Table 2: Pharmacokinetic parameters of letrozole after a single 2.5-mg oral dose of test and reference formulations in healthy Chinese female and male subjects (N = 13). Data are mean ± SD.
*Jiangsu Hengrui Medicine Co., Ltd., batch number 09112856 †Femara® letrozole 2.5 mg tablets, Novartis Pharmaceuticals Corporation East Hanover, batch number F4240. |
|
| Â |
| Bioequivalence analysis |
| Â |
| Bioequivalence analysis for the test formulation versus reference formulation was evaluated by the statistical comparisons of the primary PK parameters (Cmax and AUClast) using WINNONLIN software (BE Wizard). The BE analysis results including all the 13 subjects of each treatment were presented in Table 3. The 90% confidence interval (CI) of Cmax and AUClast for the test formulation was within the ranges of 91.97% - 111.30% and 84.97% - 102.31% of that for the reference formulation, respectively, which met the BE criteria. |
| Â |
|
|
Table 3: Comparison of 90% Cls of parameters for 2 formulations (test*/reference†) in healthy Chinese subjects (N=13). |
|
| Â |
| Discussion |
| Â |
| Metastatic breast cancer is a treatable but incurable disease [12]. In last year, aromatase inhibitors dominated the strategy of hormone treatment for breast cancer. Currently the orally-administered, third-generation aromatase inhibitors like the formulation tested are highly active and generally well tolerated. Here we showed that elimination half-life was observed between 27~121 hours for the test formulation and 39~130 hours for the reference formulation, respectively. That means the test drug’s half-life is very long [13]. Because of the plasma samples collected only up to 72 hours in this pilot study, AUC extrapolations were greater than 25% for all the subjects, AUC0-inf values are not reliable. |
| Â |
| As following log-transformation, the primary parameters AUClast and Cmax were analyzed separately by analysis of variance (ANOVA), including terms for sequence, subject (within sequence), period and formulation. Point estimates and associated 90% confidence intervals were constructed for the difference of test (T)-reference (R). The point and interval estimates on the log-scale were back-transformed to give estimates for the ratio of T:R. The 90% confidence intervals for the ratios T:R formed the basis for assessment of bioequivalence. For each primary parameter, the formulations are considered bioequivalent if the 90% confidence interval for T:R is completely contained within the range of 80-125%. |
| Â |
| The results indicate that the test and reference formulations are bioequivalent based upon the findings that the 90% confidence intervals for T:R AUClast and Cmax are contained within the range of 80-125%. For AUClast the 90% confidence interval for T:R is 84.97% to 102.31%. For Cmax the 90% confidence interval for T:R is 91.97% to 111.30%. |
| Â |
| |
| References |
| Â |
- Cancer Information Section (2002) World Health Organization, GLOBOCAN.
- Piccart MJ (1998) The changing landscape of breast cancer clinical research. ESMO-Award Lecture, ECCO-9, Hamburg. 18 September 1997. Ann Oncol 9: 133-138.
- Sabnis G, Brodie A (2011) Adaptive changes results in activation of alternate signaling pathways and resistance to aromatase inhibitor resistance. Mol Cell Endocrinol 340: 142-147.
- Shaw HS, Ellis MJ (2002) Letrozole in the treatment of breast cancer. Expert Opin Pharmacother 3: 601-617.
- Chen S, Masri S, Wang X, Phung S, Yuan YC, et al. (2006) What do we know about the mechanisms of aromatase inhibitor resistance? J Steroid Biochem Mol Biol 102: 232-240.
- Visram H, Kanji F, Dent SF (2010) Endocrine therapy for male breast cancer: rates of toxicity and adherence. Curr Oncol 17: 17-21.
- Doyen J, Italiano A, Largillier R, Ferrero JM, Fontana X, et al. (2010) Aromatase inhibition in male breast cancer patients: biological and clinical implications. Ann Oncol 21: 1243-1245.
- World Medical Association (WMA) Declaration of Helsinki (2008) Ethical Principles for Medical Research Involving Human Subjects. Adopted by the 18th WMA General Assembly, Helsinki, Finland, June 1964, and amended by the 59th WMA General Assembly, Seoul, Korea.
- Guideline for Good Clinical Practice. International Conference on Harmonisation–World Health Organization ICH topic E 6 (R1).
- Guidelines for good Clinical Practice (2010) State Food and Drug Administration.
- US Food and Drug Administration (FDA). Draft Guidance on Letrozole
- McArthur HL, Morris PG (2010) Aromatase inhibitor strategies in metastatic breast cancer. Int J Women Health 1: 67-72.
- Sioufi A, Sandrenan N, Godbillon J, Trunet P, Czendlik C, et al. (1997) Comparative bioavailability of letrozole under fed and fasting conditions in 12 healthy subjects after a 2.5 mg single oral administration. Biopharm Drug Dispos 18: 489-497.
|
| Â |
| Â |