| Research Article |
Open Access |
|
Poonam M More1*, Rishikesh V Antre1, Tushar T Shelke1, Madhavi M Mutha1,
Sahar M Badr2, Hassan M Eisa2 and Pradeep V Kore1 |
| 1Medicinal Chemistry Research Laboratory, JSPM’S Charak College of Pharmacy and Research, Pune, Maharashtra, India |
| 2Department of Organic Chemistry, Faculty of Pharmacy, Mansoura, Egypt |
| *Corresponding author: |
Poonam M More
Medicinal Chemistry Research Laboratory
JSPM’S Charak College of Pharmacy and Research
Pune, 412 207, Maharashtra, India
E-mail: rishiantre@gmail.com |
|
| Â |
| Received September 26, 2012; Published October 27, 2012 |
| Â |
| Citation: More PM, Antre RV, Shelke TT, Mutha MM, Badr SM, et al. (2012) Docking Studies of Benzimidazole Derivatives as Coenzyme-A Carboxylase (ACCase) Inhibitor. 1:439. doi:10.4172/scientificreports.439 |
| Â |
| Copyright: © 2012 More PM, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| Â |
| Abstract |
| Â |
| In these continuing efforts towards the development of disease modifying treatment for microbial diseases, we are targeting acetyl coenzyme-A carboxylase (ACCase) and it is essential for pathogen survival. In present study we reported the docking studies of substituted benzimidazole derivatives with acetyl coenzyme-A carboxylase inhibitor. |
| Â |
| Keywords |
| Â |
| Acetyl coenzyme-A carboxylase inhibitor (Accase); Benzimidazole; Antimicrobial activity |
| Â |
| Introduction |
| Â |
| Benzimidazole [1] is a heterocyclic aromatic organic compound. This bicyclic compound consists of the fusion of benzene and imidazole. Benzimidazole derivatives play important role in medical field. Benzimidazole derivatives have found the appreciation in diverse therapeutic areas including antimicrobial activity [2,3], the activity against several viruses such as HIV [4,5], antiallergic [6,7], antioxidant [8], antihistaminic [9], antitubercular [10], anti-diabetic [11], antitumor [12] activity. |
| Â |
| Many researchers have concentrated on the development of small molecules of ACCase inhibitor as antimicrobial activity. Several bacteria such as gram positive, gram negative bacteria are responsible for the microbial diseases [13]. ACCase catalyzes the first step of fatty acid biosynthesis. This reaction involves three functional components all related to a carboxybiotinyl intermediate. A biotinyl domain is covalently attached biotin prosthetic group between the active sites of a biotin carboxylase and a carboxyl transferase. In Escherichia coli, the three components reside in separate subunits; a biotinyl domain is the functional portion of one of these, biotin carboxyl carrier protein (BCCP) [14,15]. |
| Â |
| All biotin-dependent enzymes [16] share a common feature: a biotinyl domain that transfers an activated carboxyl group to a transcarboxylase center. ACCase is a multicomponent biotin enzyme present in all animals, plants and bacteria. It catalyzes one of the regulated steps (the first committed step) in the biosynthesis of longchain fatty acids, namely the biotin-dependent carboxylation of acetyl- CoA to form malonyl-CoA [17,18]. Activation by an acyl derivative of coenzyme A is observed for pyruvate carboxylases isolated from a wide range of organisms [19,20]. The enzyme is widespread in higher plants [21], algae and many kinds of bacteria, and plays an anaplerotic role by replenishing C4-dicarboxylic acid to the tricarboxylic acid cycle [22-24]. In C4 plants such as maize and sugarcane and in crassulacean acid metabolism (CAM) plants such as pineapple and cactus, phosphoenolpyruvate carboxylase (PEPC) is a key enzyme in the xation of atmospheric CO2. PEPC from Escherichia coli is an allosteric enzyme which is activated by ACCase or fructose 1, 6-bisphosphate and inhibited by L-aspartate. Although the crystal structure of E. coli PEPC complexed with L-aspartate [25] was determined in 1999, the reaction mechanism for PEPC has not been determined, because both substrate analogue and divalent metal ion were not complexed with the enzyme within the crystal [21]. Docking studies of the title compounds were performed on biopredicta>>VLife MDS 4.0 using grid-based docking method to ascertain antimicrobial activity. Therefore, ACCase inhibitor is selected as a biological target for carrying out the docking study of title compounds. The crystal structure of ACCase was obtained from protein data bank, opened in MDS sheet, saved by removing water molecule and used further for docking purpose. |
| Â |
| Materials and Methods |
| Â |
| Experimental method |
| Â |
| Data preparation: The acetyl coenzyme-A carboxylase inhibitor (3JZI) with substituted benzimidazole were collected from the reported literature. The biological activity data was collected from literature [15]. The database of molecular docking study consisted of 3JZI with 24 ligand molecules. |
| Â |
| Model building: The current study was performed using the following programs: VLife MDS 4.0 which GA batch docking. The structures of substituted benzimidazole were drowning in Vlife Engine >>tools>>draw 2D molecule and molecule was saved in Dot Mol format [26]. |
| Â |
| The 2D structures of the compounds were built and then converted into the 3D with the help of VLife MDS 4.0 software. The 3D structures were then energetically minimized up to the rms gradient of 0.01 using Merck Molecular Force Field (MMFF). By using cavity determination option of software, cavities of enzyme were determined. The cavities in the receptor were mapped to assign an appropriate active site, the basic feature used to map the cavities was the surface mapping of the receptor and identifying the geometric voids as well as scaling the void for its hydrophobic characteristics. Hence, all the cavities that are present in receptor are identified and ranked based on their size and hydrophobic surface area. Cavity no. 1 is selected for docking. The active site for docking was defined as all atoms within 5 A° radius. Using biopredicta>>tool>>docking>> batch grid docking. Batch docking shows browsing of receptor, ligand (molecule), and the result generated was saved in output file. Molecules saved in output file as a docked ligand format with proper conformation and further used to check binding interactions. Result generated was saved as log file in output folder. |
| Â |
| For checking binding interaction, first receptor structure was opened in MDS followed by compound which was saved as ligand dock file. From tool option clicked on merge molecule so that compound and receptor is merged together. From biopredicta tool edited this complex and selected ligand and receptor structure to check their interactions. |
| Â |
| Result and Discussion |
| Â |
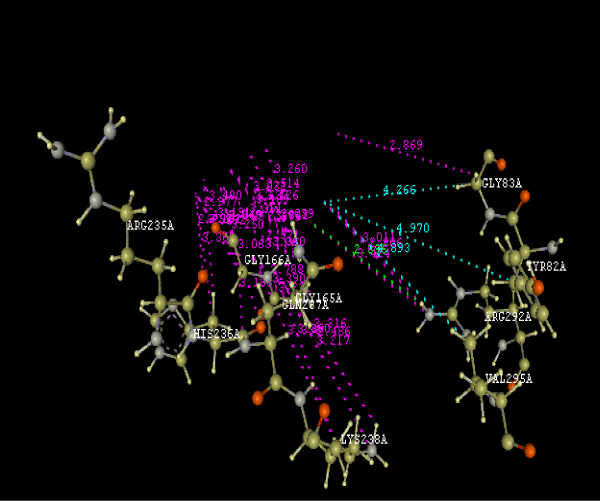
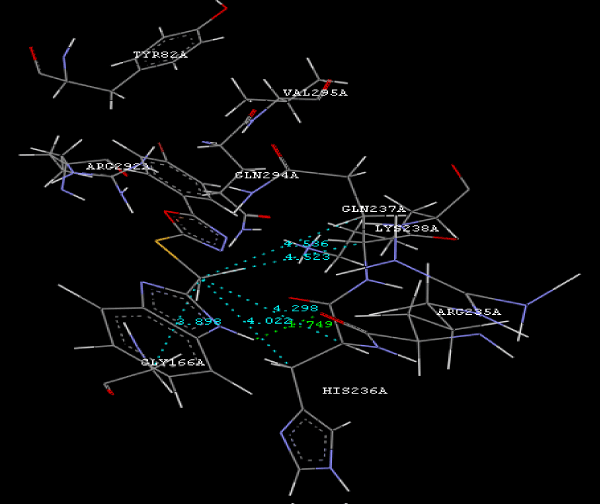
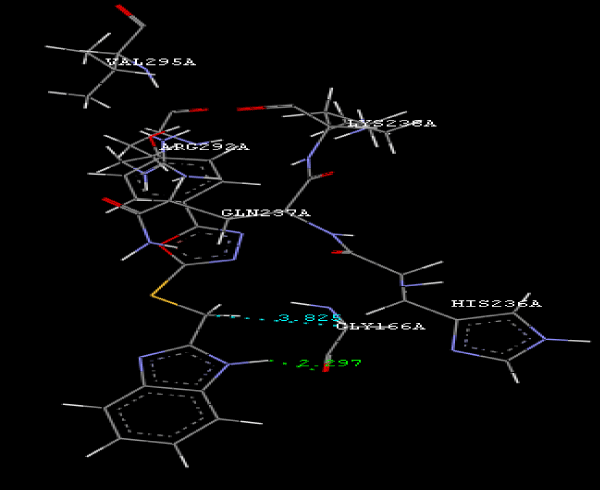
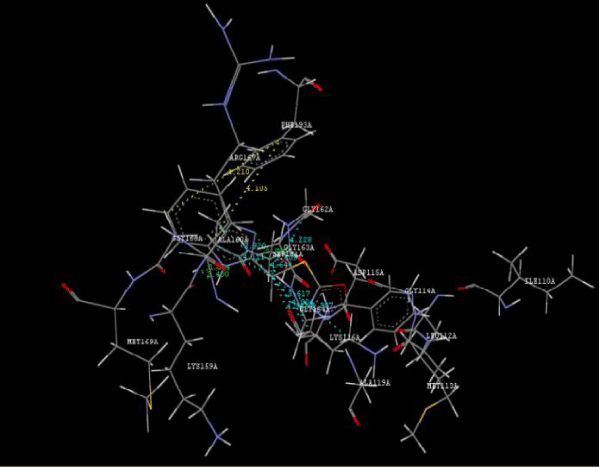
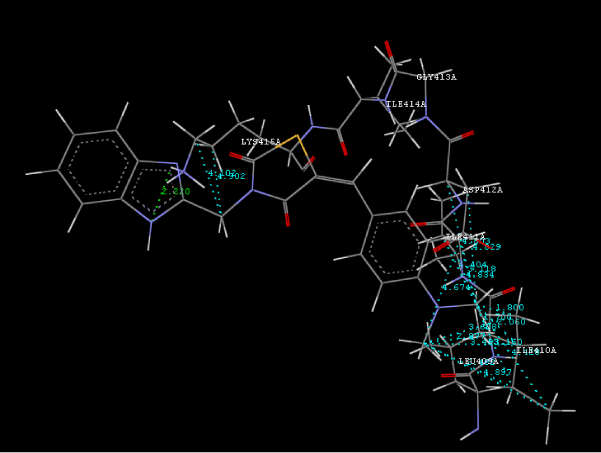
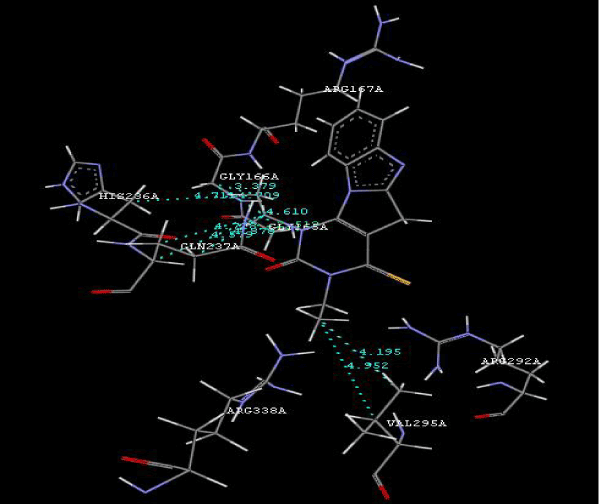
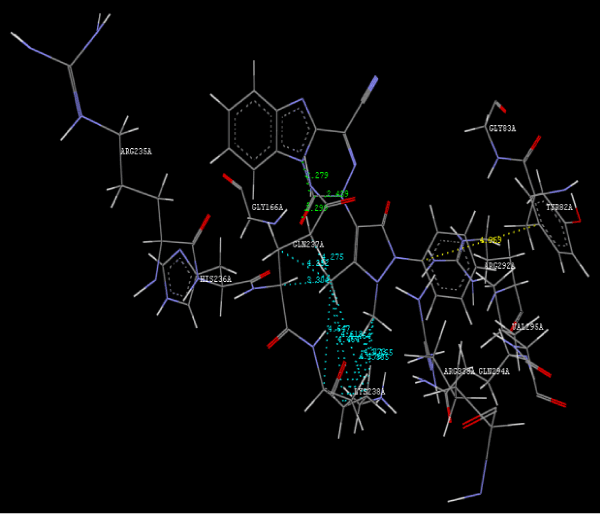
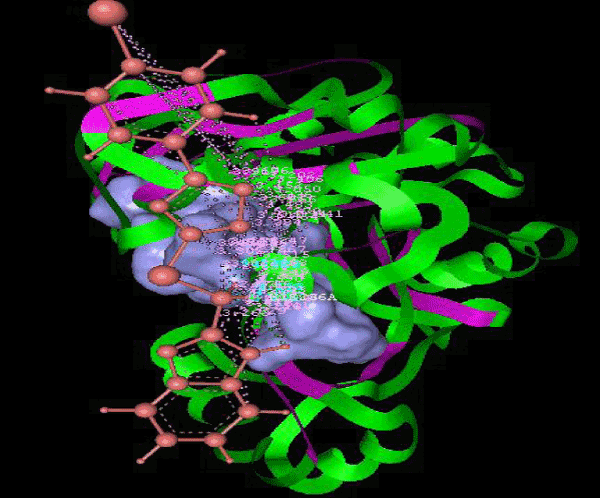
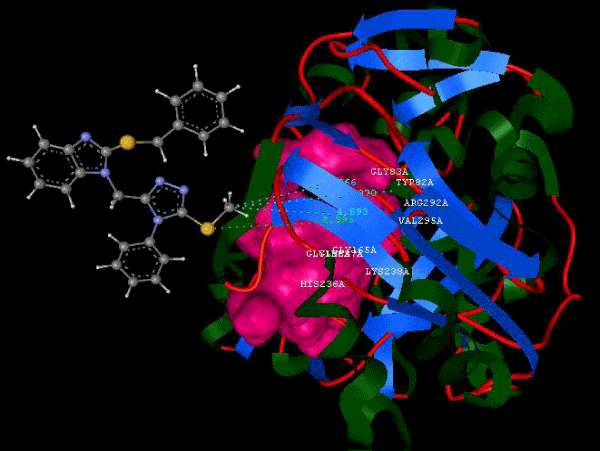
| Focused compound screening libraries are commonly used to improve the efficiency and productivity of early drug discovery efforts. Traditionally each compound in a focused library is selected based upon structural and physical properties that will increase its probability of having activity for one specific target. In this study we have extended this approach in order to identify potential multi-ligands as inhibitors. Following are the active residues involved in docking (Figures 1-3) (Table 1). |
| Â |
|
|
Figure 1a: Active amino acid residue of 3JZI involved in docking with substituted benzimidazole ligands. |
|
| Â |
|
|
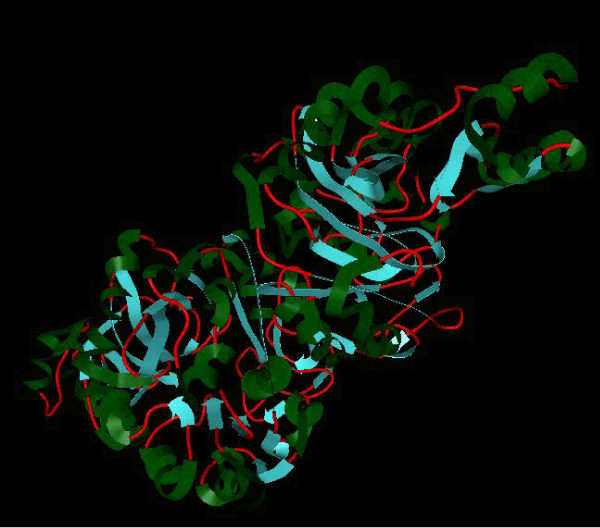
Figure 1b: Structure of PDB (3JZI) assorted view. |
|
| Â |
|
|
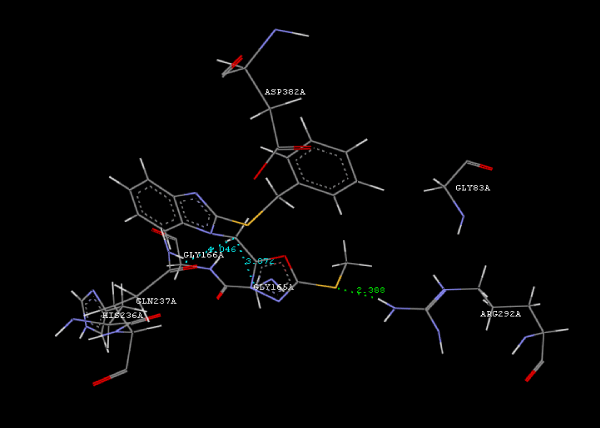
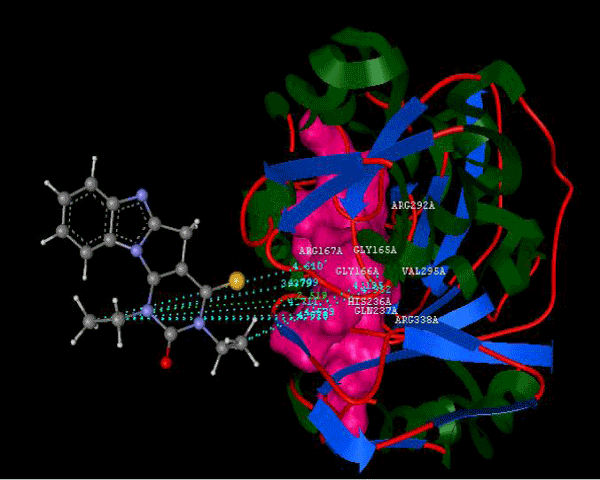
Figure 2a: Hydrogen bonding and hydrophilic Interaction of 5B ligand molecule. |
|
| Â |
|
|
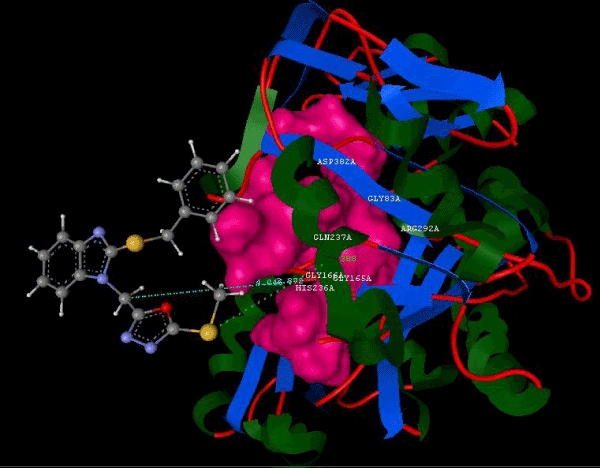
Figure 2b: Hydrogen bonding and hydrophilic Interaction of 5E ligand molecule. |
|
| Â |
|
|
Figure 2c: Hydrogen bonding and hydrophilic interaction of 7A ligand molecule. |
|
| Â |
|
|
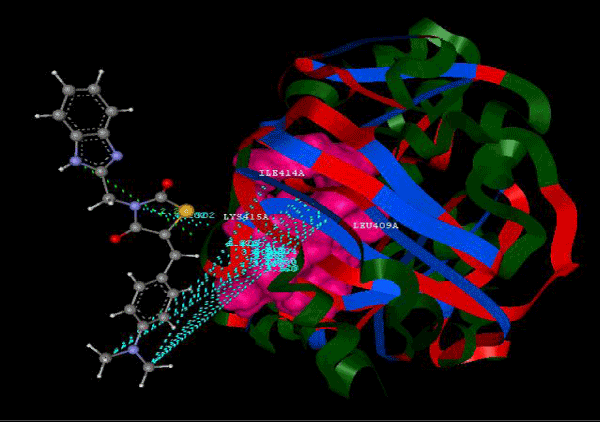
Figure 2d: Hydrogen bonding and hydrophilic interaction of 9d ligand molecule. |
|
| Â |
|
|
Figure 2e: Hydrogen bonding and hydrophilic interaction of 12B ligand molecule. |
|
| Â |
|
|
Figure 2f: Hydrogen bonding and hydrophilic interaction of 15A ligand molecule. |
|
| Â |
|
|
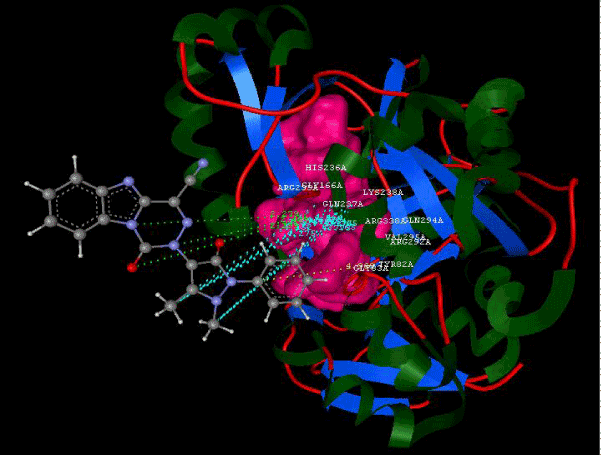
Figure 2g: Hydrogen bonding and hydrophilic interaction of 21A ligand molecule. |
|
| Â |
|
|
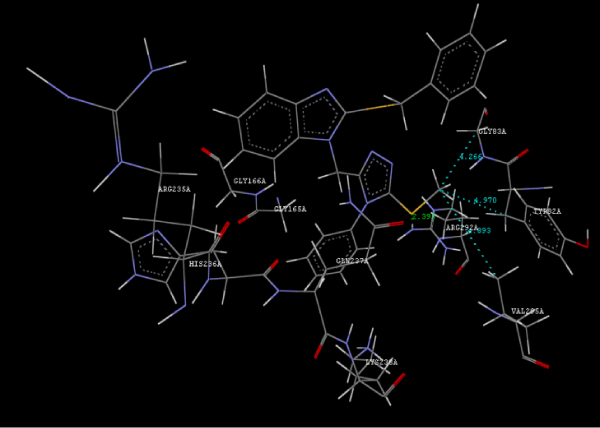
Figure 2h: Hydrogen bonding and hydrophilic interaction of 23A ligand molecule. |
|
| Â |
|
|
Figure 3a: Ligand molecule 5B docked in cavity no.1 of PDB (3JZI). |
|
| Â |
|
|
Figure 3b: Ligand molecule 23A docked in cavity no.1 with assorted representation of PDB (3JZI). |
|
| Â |
|
|
Figure 3c: Ligand molecule 12B docked in cavity no.1 with assorted representation of PDB (3JZI). |
|
| Â |
|
|
Figure 3d: Ligand molecule 15A docked in cavity no. 1 with assorted representation of PDB (3JZI). |
|
| Â |
|
|
Figure 3e: Ligand molecule 7a docked in cavity no.1 of PDB (3JZI). |
|
| Â |
|
|
Figure 3f: Ligand molecule 9D docked in cavity no.1 of PDB (3JZI). |
|
| Â |
|
|
Figure 3g: Ligand molecule 21A docked in cavity no.1 with assorted representation of PDB (3JZI). The figures 2a-2h shows the Hydrogen bond interaction in green color and the hydrophobic interaction blue color. The figures 3a-3g shows the ligand molecule docked in cavity no.1 with the receptor. The logP value (Table 1) of ligands 9E, 12A, 15A shows the very less as compared with other ligands, because 9E, 12A and 15A contains two carbonyl group substitution. Although, the substituted elucidated, molecular docking experiments indicates that ligand binds at active site residue only predicted by Biopredicta >>VLifeMDS 4.0 as shown in figure 1a. |
|
| Â |
|
|
Table 1: Interaction energy values (Kcal/mol) between ACCase and ligand molecule, log P values for ligand molecules. |
|
| Â |
| Conclusion |
| Â |
| To our knowledge, this is first report correlating the antimicrobial activity with acetyl coenzyme A carboxylase inhibitors protein and describing the binding site of protein. From protein ligand interaction as structure base drug design we reported that ARG292A, TYR82A, GLY83A, GLY165A, GLY166A, HIS236A, GLN 237A, LYS238A, VAL295A are involved in molecular docking. In conclusion, we have also reported a novel series of benzimidazole scaffold as 9B, 9D, 15A ligand that maintains the important binding site in the acetyl coenzyme-A carboxylase inhibitors by exhibiting good mol dock score. |
| Â |
| Acknowledgement |
| Â |
| The authors gratefully acknowledge to the Founder Secretary Prof. T. J. Sawant, JSPM’s group of institution for providing the constant support during research project work. |
| Â |
| |
| References |
| Â |
- Vijayakumar K, Ahamed AJ (2010) Synthesis, anti-tumor, anti-diabetic, and anti-asthmatic activitives of some novel benzimidazole derivatives. J Chem Pharma Res 2: 215-224.
- Ozden S, Atabey D, Yildiz S, Göker H (2005) Synthesis and potent antimicrobial activity of some novel methyl or ethyl 1H-benzimidazole-5-carboxylates derivatives carrying amide or amidine groups. Bioorg Med Chem 13: 1587-1597.
- Sztanke K, Pasternak K, Sidor-Wojtowicz A, Truchlinska J, Jozwiak K (2006) Synthesis of imidazoline and imidazo[2,1-c][1,2,4]triazole aryl derivatives containing the methylthio group as possible antibacterial agents. Bioorg Med Chem 14: 3635-3642.
- Porcari AR, Devivar RV, Kucera LS, Drach JC, Townsend LB (1998) Design, synthesis, and antiviral evaluations of 1-(substituted benzyl)-2-substituted-5,6-dichlorobenzimidazoles as nonnucleoside analogues of 2,5,6-trichloro-1-(beta-D-ribofuranosyl)benzimidazole. J Med Chem 41: 1252-1262.
- Roth T, Morningstar ML, Boyer PL, Hughes SH, Bukheit RW, et al. (1997) Synthesis and biological activity of novel nonnucleoside inhibitors of HIV-1 reverse transcriptase. 2-aryl substituted benzimidazoles. J Med Chem 40: 4199-4207.
- Ahmadi A, Nahri-Niknafs B (2011) Synthesis, characterization of some novel benzimidazole derivatives of 1-bromo-2, 4-dinitrobenzene and their antifungal activities. E-Journal of Chemistry 8: S85-S90.
- Nakano H, Inoue T, Kawasaki N, Miyataka H, Matsumoto H, et al. (1999) Synthesis of benzimidazole derivatives as antiallergic agents with 5-lipoxygenase inhibiting action. Chem Pharm Bull (Tokyo) 47: 1573-1578.
- Can-Eke B, Puskullu MO, Buyukbingol E, Iscan M (1998) A study on the antioxidant capacities of some benzimidazoles in rat tissues. Chem Biol Interact 113: 65-77.
- Goeker H, Ayhan-Kilcigil G, Tuncbilek M, Kus C, Ertan R, et al. (1999) Synthesis and antihistaminic H-1 activity of 1,2,5(6)- Trisubstituted benzimidazoles. Heterocycles 51: 2561-2573.
- Camacho J, Barazarte A, Gamboa N, Rodrigues J, Rojas R, et al. (2011) Synthesis and biological evaluation of benzimidazole-5-carbohydrazide derivatives as antimalarial, cytotoxic and antitubercular agents. Bioorg Med Chem 19: 2023-2029.
- Senten K, Van der Veken P, De Meester I, Lambeir AM, Scharpé S, et al. (2003) Design, synthesis, and SAR of potent and selective dipeptide-derived inhibitors for dipeptidyl peptidases. J Med Chem 46: 5005-5014.
- Garuti L, Roberti M, Malagoli M, Rossi T, Castelli M (2000) Synthesis and antiproliferative activity of some benzimidazole-4,7-dione derivatives. Bioorg Med Chem Lett 10: 2193-2195.
- Cheng CC, Shipps GW Jr, Yang Z, Sun B, Kawahata N, et al. (2009) Discovery and optimization of antibacterial AccC inhibitors. Bioorg Med Chem Lett 19: 6507-6514.
- Athappilly FK, Hendrickson WA (1995) Structure of the biotinyl domain of acetyl-coenzyme A carboxylase determined by MAD phasing. Structure 3: 1407-1419.
- Shen Y, Volrath SL, Weatherly SC, Elich TD, Tong L (2004) A mechanism for the potent inhibition of eukaryotic acetyl-coenzyme A carboxylase by soraphen A, a macrocyclic polyketide natural product. Mol Cell 16: 881-891.
- Knowles JR (1989) The mechanism of biotin-dependent enzymes. Annu Rev Biochem 58: 195-221.
- Wakil SJ, Stoops JK, Joshi VC (1983) Fatty acid synthesis and its regulation. Annu Rev Biochem 52: 537-579.
- Lane MD, Moss J, Polakis SE (1974) Acetyl coenzyme A carboxylase. Curr Top Cell Regul 8: 139-195.
- Osmani SA, Scrutton MC (1981) Activation of pyruvate carboxylase from aspergillus nidulans by acetyl coenzyme A. FEBS Let 135: 253-256.
- Scrutton MC (1978) Fine control of the conversion of pyruvate (phosphoenolypyruvate) to oxaloacetate in various species. FEBS Lett 89: 1-9.
- Matsumura H, Terada M, Shirakata S, Inoue T, Yoshinaga T, et al. (1999) Plausible phosphoenolpyruvate binding site revealed by 2.6 A structure of Mn2+-bound phosphoenolpyruvate carboxylase from Escherichia coli. FEBS Lett 458: 93-96.
- Utter MF, Kolenbrander HM (1972) Formation of oxaloacetate by CO2 fixation on P- enolpyruvate. The Enzyme 7: 117-165.
- Ausenhus SL, O'Leary MH (1992) Hydrolysis of phosphoenolpyruvate catalyzed by phosphoenolpyruvate carboxylase from Zea mays. Biochemistry 31: 6427-6431.
- Chollet R, Vidal J, O'Leary MH (1996) Phosphoenolpyruvate Carboxylase: A Ubiquitous, Highly Regulated Enzyme in Plants. Annu Rev Plant Physiol Plant Mol Biol 47: 273-298.
- Kai Y, Matsumura H, Inoue T, Terada K, Nagara Y, et al. (1999) Three-dimensional structure of phosphoenolpyruvate carboxylase: a proposed mechanism for allosteric inhibition. Proc Natl Acad Sci U S A 96: 823-828.
- Eisa HM, Barghash AM, Badr SM, Farahat AA (2010) Synthesis & antimicrobial activity of certain benzimidazole and fused benzimidazole derivatives. Ind J Chem 49B: 1515-1525.
|
| Â |
| Â |