| Research Article |
Open Access |
|
| Syed Hussain Basha* and Kazipet Naresh Kumar |
| Independent Researcher, 117- D, WWSC, Guntupalli, Ibrahimpatnam, Vijayawada - 521 241, India |
| *Corresponding author: |
Syed Hussain Basha
Independent Researcher, 117- D, WWSC, Guntupalli
Ibrahimpatnam, Vijayawada - 521 241, India
Tel: +919177247605
E-mail: hassainbasha53@gmail.com |
|
| Â |
| Received September 24, 2012; Published December 28, 2012 |
| Â |
| Citation: Hussain BS, Naresh Kumar K (2012) Ligand and Structure Based Virtual Screening Studies to Identify Potent Inhibitors against Herpes Virus Targeting gB-gH-gL Complex Interface as a Novel Drug Target. 1:566 doi:10.4172/ scientificreports.566 |
| Â |
| Copyright: © 2012 Hussain BS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. |
| Â |
| Abstract |
| Â |
| Glycoproteins gB and gH-gL are highly conserved cell entry machinery, which are involved in attachment and fusion of herpes virus to the host cell. gB is a homotrimer with structural characteristics to undergo conformational rearrangement when triggered, thus inferred to be the effector of viral fusion, whereas, gH-gL glycoprotein is a heterodimer complex proposed to be the activator of gB glycoprotein, probably through direct binding. Critical dependence of herpes virus on the formation of this gB-gH-gL complex for its entry into the host cell, making this interface a promising anti herpes drug target. Arresting this complex formation by blocking the interactions between the key residues of these glycoproteins seems to be the most promising mechanism to inhibit the viral infection. From our previous research, we identified (3-Chloro Phenyl) Methyl-3,4,5 Trihydroxybenzoate (CPMTHB) as a potent inhibitor for gH-gL heterodimer complex. In this present study, a ligand based virtual screening with a threshold of >50% similarity was performed, based on the structure of CPMTHB using ZINC database, and resulted 505 compounds were utilized to perform a structure based virtual screening on glycoproteins gB and gH-gL complex separately, targeting key residues involved in their binding activity. 31 compounds were identified as better inhibitors based on free binding energy and ADMET constraints, compared to CPMTHB. The capability of CPMTHB and 31 better compounds to disrupt gB-gH-gL complex formation was evident from our flexible and semi-flexible docking studies, suggesting the possible mode of action of these tested compounds to inhibit herpes virus, is by attenuating this complex formation, thus leaving significant evidence in support of this complex as a promising anti herpes drug target. |
| Â |
| Keywords |
| Â |
| Herpes simplex virus; gB–gH–gL complex; Virtual screening; Docking; (3-chloro phenyl) methyl-3,4,5 trihydroxybenzoate |
| Â |
| Background |
| Â |
| Herpes Simplex Virus (HSV) is a double-stranded DNA virus, belongs to Herpesviridae family which is ubiquitous, contagious, hostadapted pathogen, and its infection is one of the most common viral sexually transmitted diseases worldwide [1,2]. There are two types of HSV: HSV-1, which is traditionally associated with orofacial disease (Herpes labialis), and has emerged as a principle causative agent of genital herpes in some developed countries [1,3,4]. HSV-1 is a vital cause for the genital herpes in United States (US), and is significantly reported among the college students [1,5,6]; while HSV-2 is one of the most common sexually transmitted infections, traditionally associated with genital diseases (Herpes genitalis) [7,8], and reported to be the most obvious cause of genital ulcer disease in all regions of the world [9-12]. 22% of adults in US were found to be containing antibodies to HSV-2 in a population based study, which lead to an estimated 1.6 million new cases every year [13-14]. Both HSV-1 and 2 are highly infectious, establishes latency in neurons, and may reactivate to cause recurrent lesions. Herpes viruses make their initial contact with cells by binding heparan sulfate, which has thought to be an irreversible process, because this binding immediately triggers envelopemembrane or cell-cell fusion [15]. Fusion is achieved with the help of conserved fusion machinery components, glycoproteins gB and gH-gL complex, along with other non-conserved components. Therefore, this highly conserved fusion machinery is the most promising target in the discovery of novel anti-herpes drugs [16,17]. |
| Â |
| From our pervious investigation, we have identified CPMTHB as a potent inhibitor for gH-gL complex, based on free binding energy and pharmacological properties [18]. In this present study conducted, we have performed a ligand based virtual screening based on the structure of CPMTHB, to identify much better binding compounds with the constraints of ADMET properties. The 505 compounds thus found were utilized to perform a structure based virtual screening for gB and gH-gL glycoproteins separately, targeting the active residues in their binding activity. 31 compounds which showed lesser free binding energy than CPMTHB were further analyzed for their capability to disrupt this complex, by performing flexible and semi-flexible docking studies. |
| Â |
| Structural aspects of glycoproteins gB & gH-gL |
| Â |
| gB glycoprotein is a spike shaped homotrimer, with approximate dimensions of 85 Ã…, 80 Ã… and 160 Ã…, containing 904 residues in each promoter among the trimer. Each promoter is composed of five domains. Domain I (base) is a continuous poly peptide composed of ILE154 to VAL363 amino acid residues, Domain II (middle) is discontinuous two segmented chain from TYR142 to ASN153 and CYS364 to THR459, Domain III (core) consists of three discontinuous segmented from PRO117 to PRO133, SER500 to THR572, and ARG661 to THR 669, Domain IV (crown) comprises two discontinuous segments, residues ALA111 to CYS116 and CYS573 to SER660. Domain V (arm) residues, PHE670 to ALA725, stretches from top to bottom of the molecule as a long extension. |
| Â |
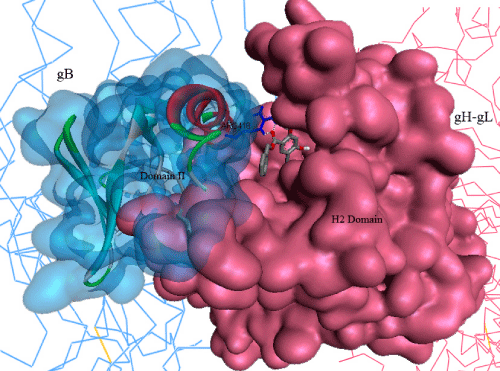
| gH–gL is a boot shaped heterodimer complex, which is ~80 Å high and ~70 Å long, contains residues Gly48–Pro803 of gH, followed by a C-terminal hexahistidine tag and residues Gly20–Asn224 of gL. gH has three distinct domains: the N-terminal domain that binds gL (domain H1), extended from GLY48 to PRO327, is located in the upper part of the gH–gL boot and consists of sub-domains H1A and H1B, which are connected by a linker (residues Gly116–Pro136). The central helical domain (domain H2) is globular and mostly helical structurally, and extended from ASN332 to PHE644. The C-terminal domain (domain H3) is located at the toe end of the boot and extended from residues VAL645 to PRO797. The complete structural features of the glycoproteins gB [PDB: 2GUM] and gH-gL complex [PDB: 3M1C] have been described in detail elsewhere [16,17], by the depositors of these glycoprotein crystal structures to the Protein Data Bank. |
| Â |
| From our previous study, we have identified LYS 435, ARG 418, GLN 438, PRO 439, LEU 399, GLU 401 and GLY 437 of gB and VAL 342, GLU 347, SER 349, TYR 355, SER 388, ASN 395, HIS 398 and ALA 387 of gH-gL complex are active in their binding activity [18]. These key residues have been defined as the active sites for gB and gHgL glycoproteins, respectively, in the present study. |
| Â |
| gB-gH-gL complex as a therapeutic drug target |
| Â |
| Glycoproteins B, H & L has been well reported as the most conserved cell entry machinery [15,19]. The activation of gB possibly through direct binding of gH-gL heterodimer complex has been presumed to be the key step for the viral entry into the host cell [16]. Moreover, the structural features of gB strongly suggests its critical activity as a key effector for viral fusion [17]. Inhibition of its structural rearrangement seems to be the most promising mechanism to inhibit viral entry into the host cell. Atanasiu et al. [20] proposed Domain II of gB consists the key amino acid residues, which are involved in its binding activity with gH-gL complex, and most possibly the key domain responsible for structural rearrangements in gB, when triggered by direct binding of gH-gL, whereas, Chowdary TK et al. [16] proposed a gB binding site in H2 domain of gH-gL complex. From our previous in-silico investigations, we have identified LYS 435, ARG 418, GLN 438, PRO 439, LEU 399, GLU 401, and GLY 437 of gB and VAL 342, GLU 347, SER 349, TYR 355, SER 388, ASN 395, HIS 398, and ALA 387 of gHgL complex are active in their binding activity [18]. Moreover, all the published ethno compounds with proven in-vitro, in-vivo activity against herpes we tested in our previous study, showed strong binding activity with key residues in gH-gL complex, suggesting their possibility to inhibit herpes infection by attenuating gB-gH-gL complex formation [18]. Thus, this complex formation interface seems to be the most promising drug target for designing inhibitors against herpes, which remained unexplored so far. |
| Â |
| Development of gB-gH-gL inhibitors |
| Â |
| Entry of the herpes virus into the host cell is critically depended on the activation of gB, which is presumed to be happening by the direct binding of gH-gL heterodimer complex [16,17]. Thus, blocking the key residues involved in this binding activity seems to be a promising mode of action to be chosen for designing drug candidates against herpes virus. Currently established Acyclovir (9-[2-hydroxymethyl] guanine) is a deoxyguanosine triphosphate (dGTP) analog, whose mode of action is to competitively inhibit viral DNA polymerase of HSV [21,22]. However, there is every possibility of this nucleoside analog to interfere with normal cellular replication, whose consequences are unknown. On the other hand, reports of herpes resistance to acyclovir for its recurrents and side effects urges for novel inhibitors with higher selectivity for this virus inhibition [23]. |
| Â |
| Results and Discussion |
| Â |
| In this work, different in-silico approaches were applied to virtually screen for potential inhibitors targeting gB-gH-gL complex formation interface of Herpes simplex virus. As it is established that structurally similar compounds have same pharmacological features, we have performed a ligand based virtual screening, based on the structure of CPMTHB using ZINC database, to identify its structurally similar compounds. 505 compounds thus found with the threshold of >50% similarity were used to perform structure based virtual screening on gB and gH-gL glycoproteins, separately targeting the active residues involved in their binding activity using Arguslab 4.0 |
| Â |
| Arguslab 4.0 was run to rank these 505 compounds based on their free binding energies, to screen for the molecules that potentially binds gB and gH-gL glycoproteins. Binding affinity calculations for all 505 compounds were carried out using Genetic algorithm implemented in Arguslab 4.0. We then selected 31 compounds with stronger binding affinity for gH-gL heterodimer complex than CPMTHB (Table 1). Higher binding affinity compared to CPMTHB with good pharmacological properties is the basis for selecting these 31 compounds. Only these 31 compounds results will be discussed in further for gB and gH-gL complex, in detail. |
| Â |
|
|
Table 1: The docking simulation results of compounds towards gB and gH-gL complex. |
|
| Â |
| Structure based virtual screening |
| Â |
| One of the possible modes of action we hypothesized for these compounds is attenuating the gB-gH-gL complex formation, thus inhibiting the viral entry into the host cell in subsequent steps. In order to explore the possibility of these selected compounds to attenuate this complex formation, we carried out structure based virtual screening for gB and gH-gL, targeting the key residues involved in their active complex formation. The results will be discussed in further in two sections. |
| Â |
| Inhibitors bound to gH-gL active site: 505 compounds obtained from the ZINC database were virtually screened onto the gH-gL glycoprotein targeting its active residues. All the compounds analyzed were successfully docked with a binding energy range of -13.4 to +11.2 Kcal/mol, with the exceptions of compounds, ZINC06045419 and ZINC06641550, which are unable to dock into the target binding site, whereas compounds, ZINC40883455 and ZINC03249881, showed the highest and least binding affinity of -13.4 and +11.2 Kcal/mol, respectively. |
| Â |
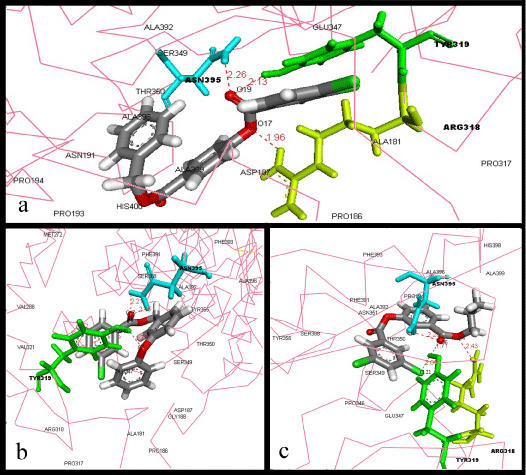
| A total of 31 compounds with higher binding affinities for gH-gL than that of CPMTHB were tabulated (Table 1). Top three compounds with the strongest affinity for the gH-gL binding site were ZINC40883455, ZINC01936038 and ZINC10154846, which showed binding energy of -13.4, -12.2 and -12.2 Kcal/mol, respectively. The best binder was compound ZINC40883455 (-13.4 Kcal/mol). This compound (Figure 1a) formed three hydrogen bonds and several Van der Waals (VDW) interactions with residues inside the gH-gL binding site. Three hydrogen bonds are formed between ARG318, TYR319 and ASN395, with O17 and O19 atoms of the compound. In addition, several VDW interactions are formed between compound ZINC40883455 and residues PRO186, ALA181, ASP187, GLU347, SER349, ASN191, PRO193, PRO194, THR350, PRO317, ALA392, ALA396, ALA399, and HIS400. |
| Â |
|
|
Figure 1: Best binders of gH-gL: Interactions between a) ZINC40883455 b) ZINC01936038 c) ZINC10154846, and the residues of gH-gL active site. The ligand and the hydrogen bond forming residues are shown in stick coloured by element and amino acid, respectively, whereas the protein was represented in ca-wire labelled with the residues which are involved in VDW interactions with ligand. |
|
| Â |
| Second best binder, followed by ZINC40883455, is compound ZINC01936038 (-12.2 Kcal/mol), which formed three hydrogen bonds between ASN395 and TYR 319, with O7 & O17 atoms of the compound, along with good VDW interactions with amino acid residues TYR355, PHE393, SER388, ALA392, ALA396, PHE391, MET272, PRO346, THR350, SER349, GLU347, VAL321, GLY188, ASP187, ALA181, PRO186, PRO317, and ARG318 (Figure 1b). |
| Â |
| As for the compound ZINC10154846, the third best binder with binding energy of -12.2 Kcal/mol, showed the highest hydrogen bonds formation amongst these top three compounds, which is four hydrogen bonds, among these bonds three are formed by O6 and CL22 with TYR319 and ASN395, whereas O4 formed a bond with ARG318. This compound also showed several VDW interactions with HIS398, ALA399, ALA396, PHE393, ALA392, PHE391, PRO194, ASN351, THR350, SER388, TYR355, SER349, PRO346, and GLU347 (Figure 1c). |
| Â |
| From our previous research, we have identified that VAL 342, GLU 347, SER 349, TYR 355, SER 388, ASN 395, HIS 398 and ALA 387 amino acid residues of gH-gL complex are significantly important in its binding activity with glycoprotein gB. Three best binders among the analyzed compounds were found to be creating one or more hydrogen bonds with ASN395. Moreover, ARG318 and TYR319 which formed hydrogen bonds with the compounds were also found to be significant role players in ligand binding with gH-gL binding site in our previous research [18]. Our results indicate that these three compounds can block the gH-gL binding activity with gB by binding to its critical residues. |
| Â |
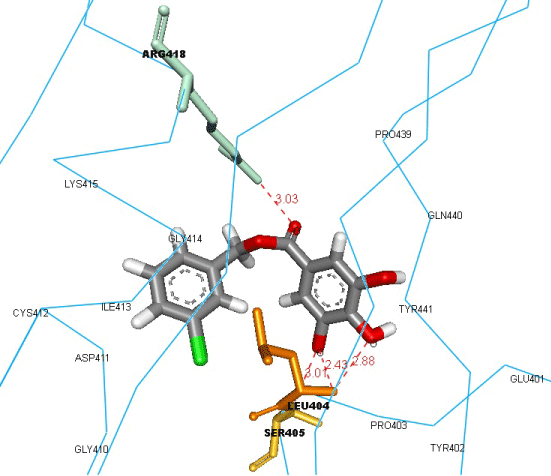
| Inhibitors bound to gB active site: Firstly, we performed a semiflexible docking for CPMTHB compound, targeting the active amino acid residues of gB glycoprotein to check its binding affinity. It was successfully docked with a binding energy of -9.8 Kcal/mol (Figure 2), and found to be forming three hydrogen bonds with ARG418, LEU404 and SER405, along with VDW interactions with LYS415, CYS412, ASP411, ILE413, GLY410, GLY414, PRO403, TYR402, GLU401, TYR441, GLN440, and PRO439. The hydrogen bond formed with ARG418 showed CPMTHB’s capability to inhibit binding activity of gB with gH-gL complex, as this residue was found to be active in forming hydrogen bond with GLU347 of gH-gL complex in our previous study [18]. We then performed a semi-flexible docking study targeting the key residues of gB active site, to check their capability to inhibit gB glycoprotein interaction with gH-gL complex, using 31 better compounds found for the gH-gL complex, results of this semi-flexible docking simulations can be depicted from table 1 and the snapshots of the interactions, along with the structures of these 31 better compounds can be found in supplementary material 1. |
| Â |
|
|
Figure 2: Binding of CPMTHB into the active site of gB glycoprotein, and the residues involved in interactions with the ligand. The ligand and the hydrogen bond forming residues are shown in stick coloured by element and amino acid, respectively, whereas the protein was represented in ca-wire labelled with the residues, which are involved in VDW interactions with ligand. |
|
| Â |
| After this successful dock results, we performed structure based virtual screening on gB, targeting its active amino acid residues, using 505 compounds obtained from the ZINC database screen. All the compounds utilized for the screen were shown to be successfully docked with a binding energy range of -5.4 to -12.5 Kcal/mol. These compounds were ranked based on their binding energies. Compound ZINC08277636 showed the highest binding affinity of -12.5 Kcal/mol, whereas compound ZINC12661135, showed the least binding affinity of -5.4 Kcal/mol. 42 compounds which showed less than or equal to the binding energy of CPMTHB to gB active site has been shown in table 2. |
| Â |
|
|
Table 2: The compounds which showed less than or equal to the binding energy of CPMTHB with gB glycoprotein. |
|
| Â |
| Among these 42 compounds, ZINC01972391 and ZINC04799874, were found to be the common strong inhibitors for both gB and gH-gL complex, with -10.7 & -10.1 Kcal/mol binding energy for gB, and -10.6 & -10.4 Kcal/mol binding energy for gH-gL complex, respectively. As our motive is to identify potent inhibitors which can attenuate gB-gHgL complex formation, we will further discuss about these two common inhibitors only, instead of discussing about top three best binders of gB. |
| Â |
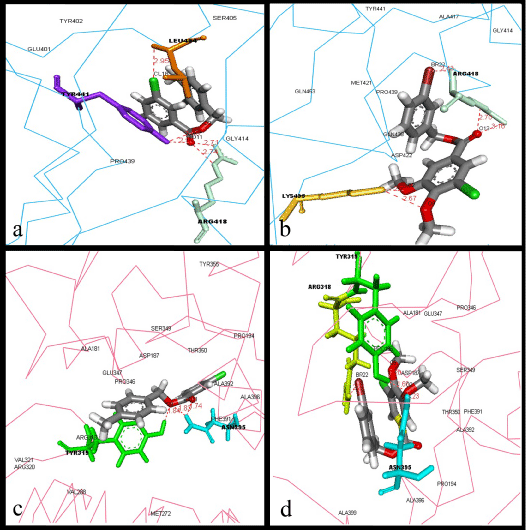
| Compound ZINC01972391 (Figure 3a) formed five hydrogen bonds between O9 & O11, CL18 and LEU404, TYR441 and ARG418. VDW interactions have been observed between the compound and the amino acid residues GLU401, TYR402, SER405, GLY414, and PRO439. The second potential inhibitor, compound ZINC04799874 (Figure 3b), also formed five hydrogen bonds between O2, O9, O13 and BR22 with LYS435 and ARG418, along with VDW interactions with GLN438, TYR441, GLY414, PRO439, GLN453, ALA417, MET421, and ASP422. |
| Â |
|
|
Figure 3: Best binders for both gB & gH-gL: Interactions between a) ZINC01972391 with residues of gH-gL active site. b) ZINC04799874 with residues of gH-gL active site. c) ZINC01972391 with residues of gB active site. d) ZINC04799874 with residues of gB active site. The ligand and the hydrogen bond forming residues are shown in stick coloured by element and amino acid, respectively, whereas the protein was represented in ca-wire labelled with the residues, which are involved in VDW interactions with ligand. |
|
| Â |
| When these two compounds were analyzed for their ability to bind with gH-gL complex, compound ZINC01972391 (Figure 3c) formed three hydrogen bonds, among which two bonds were formed between O9 & O11 and ASN395, whereas the third bond was formed between O9 and TYR319. VDW interactions have been observed between the compound and the amino acid residues MET272, VAL288, VAL321, ARG320, ARG318, PHE391, ALA396, ALA392, PRO346, GLU347, PRO194, TYR355, THR350, SER349, ASP187 and ALA181. The second potential inhibitor, compound ZINC04799874 (Figure 3d), formed four hydrogen bonds, among which three bonds formed between O2, O9 and TYR319 and ASN395, whereas the fourth bond was formed between BR22 and ARG318, along with VDW interactions with PRO194, ALA181, PRO186, ASP187, SER349, THR350, GLU347, PRO346, ALA392, PHE391, ALA396, and ALA399. The structures of the best binders for gB and gH-gL can be depicted from figure 4. |
| Â |
|
|
Figure 4: Best binders of gH-gL: Interactions between a) ZINC40883455 b) ZINC01936038 c) ZINC10154846, and the residues of gH-gL active site. The ligand and the hydrogen bond forming residues are shown in stick coloured by element and amino acid, respectively, whereas the protein was represented in ca-wire labelled with the residues which are involved in VDW interactions with ligand. |
|
| Â |
| From our previous study, we have identified that LYS 435, ARG 418, GLN 438, PRO 439, LEU 399, GLU 401 and GLY 437 of gB protein are significantly important in its binding activity with glycoprotein gH-gL heterodimer complex. Our results show that compound ZINC01972391 binds to the gB binding site by forming interactions with three residues: LEU404, TYR441 and ARG418, whereas compound ZINC04799874 binds to the gB binding site by forming interactions with two residues: ARG418 and LYS435. Among these residues, ARG418 and LYS435 showed to be interacting with GLU347 and VAL342 of gHgL complex, respectively, in our previous study. On the other hand, when ZINC01972391 binds to the gH-gL binding site, it had formed interactions with two residues: TYR319 and ASN395, whereas, when compound ZINC04799874 binds to the gH-gL binding site, it has formed interactions with three residues: ARG318, TYR319 and ASN395. Among these residues, ASN395 was showed to be interacting with PRO439 of gB glycoprotein in our previous study. These results leave significant evidence of these compounds capability to attenuate the gB-gH-gL complex interface by blocking the key residues. |
| Â |
| Flexible docking results |
| Â |
| We have performed a flexible docking study to know the efficacy of CPMTHB and 31 better compounds, to disrupt the gB-gH-gL active complex by keeping the active residues of this complex formation as flexible, along with the given ligand molecule for each docking. When CPMTHB was docked into this active complex, it was found to be forming hydrogen bonding with GLU362, GLY365 and ASP367, stabilized by VDW interactions with ARG381, LEU378, ARG418 and ASP422 (Figure 5), with binding energy of -1.94 Kcal/mol. |
| Â |
|
|
Figure 5: Best binders of gH-gL: Interactions between a) ZINC40883455 b) ZINC01936038 c) ZINC10154846, and the residues of gH-gL active site. The ligand and the hydrogen bond forming residues are shown in stick coloured by element and amino acid, respectively, whereas the protein was represented in ca-wire labelled with the residues which are involved in VDW interactions with ligand. |
|
| Â |
| The binding of CPMTHB to the active complex provides significant evidence in support of our hypothesized mechanism of gB-gH-gL activation suppression, by inhibition or disruption of this complex. The binding energy results were obtained from clusters obtained for genetic algorithm run of ten models. The binding energy (-1.94 Kcal/mol) of CPMTHB to the active gB-gH-gL complex was much higher than that of segregated gB (-9.8 Kcal/mol), and gH-gL complex (-10.4 Kcal/mol), which shows thermodynamic instability of this compound to disrupt this active complex, in contrast with its high stability, when docked with segregated gB and gH-gL complex. Only notable positive result with this compound is the VDW interactions with ARG418, one of the active residues of the gB glycoprotein found to be forming hydrogen bonding with GLU347 of gH-gL from our previous study. |
| Â |
| All the 31 compounds which showed lesser binding energy than CPMTHB, towards gH-gL active site, has also been tested for their ability to disrupt this complex formation. Among the 31 compounds analyzed, no compound was found to be forming any hydrogen bonding with any of the active residues in this complex formation, with the exception of compound ZINC59323245 (Figure 6), which formed three hydrogen bonds with ARG418, along with good VDW interactions with ALA361, LEU570, ARG567, GLN623, VAL627, GLY365, LEU364, ASP422, and ARG381, with a binding energy of -0.61 Kcal/mol. However, the higher binding energy (-0.61 Kcal/mol) than that obtained from binding of this compound to the segregated gB (-7.9 Kcal/mol) and gH-gL complex (-10.7 Kcal/mol) suggests its high thermo dynamical instability. |
| Â |
|
|
Figure 6: Flexible Docking simulation result of ZINC59323245: Ligand forming hydrogen bonds with ARG418, one of the active residues of the gB glycoprotein found to be forming hydrogen bonding with GLU347 of gH-gL active site. |
|
| Â |
| These results strongly suggest that these tested compounds do not possess any significant effect on disrupting this active complex. On the other hand, the higher binding affinity of these tested compounds towards the segregated gB, gH-gL glycoproteins clearly suggests that these compounds are capable of attenuating the gB-gH-gL complex formation, by either inhibiting gB or gH-gL complex compared to their capability to disrupt the already formed active complex. |
| Â |
| Semi-flexible docking results |
| Â |
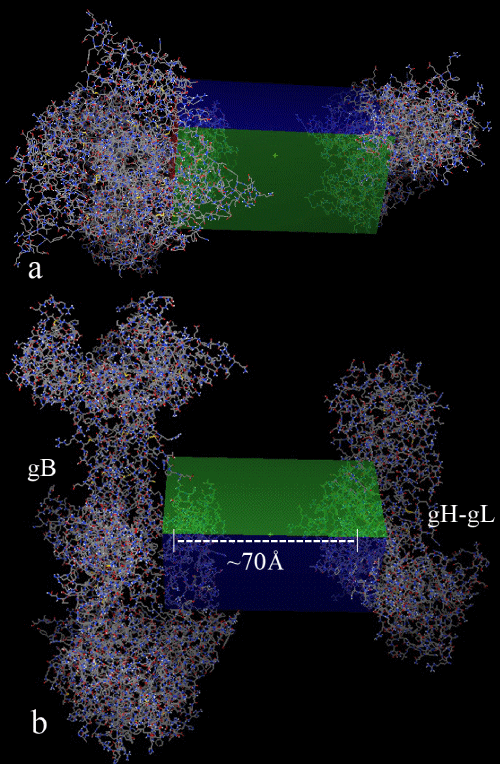
| To substantiate our hypothesis that the tested compounds are capable of attenuating the gB-gH-gL complex formation rather than their capability to disrupt the already formed active complex, and also to substantiate our previous study hypothesis that our proposed binding pocket in gH-gL complex is much preferred drug targetable, we have performed a semi-flexible docking study on CPMTHB and 31 better compounds, by keeping the gB and gH-gL complex in the same molecular axis, with a distance of ~70Ã…. This experiment was performed by taking care during the grid designing, so that only the proposed active sites in each of these glycoproteins are surrounded by the three dimensional grid space, as shown in figure 7. The results of this semi-flexible docking study have been tabulated (Table 3). |
| Â |
|
|
Table 3: Semi-flexible docking simulation results of compounds when gB and gHgL complex are kept in the same molecular axis with approx. ~70 Ã… distance. |
|
| Â |
| From the tabulated results (table 3), it is evident that all the compounds have successfully docked, either to gB or gH-gL glycoprotein, with a binding energy range between -4.4 to -7.2 Kcal/ mol. In our previous study, we proposed CPMTHB as the potent inhibitor for gH-gL complex, which has been substantiated in this present study by the successful dock of CPMTHB into the active site of gH-gL complex, with a binding energy of -5.73 Kcal/mol, even when this compound was given a choice for binding either to gB or gH-gL active site. Compound ZINC66066505, which showed to be the only promising molecule among the 31 compounds tested from the flexible docking studies, showed to be successfully docked into gH-gL active site with -4.4 Kcal/mol binding energy. Even it is the highest binding energy in this semi-flexible docking study, but it is much more thermodynamically stable compared to its binding energy of -0.61 Kcal/mol in flexible docking study, suggesting its role in inhibiting gHgL binding to gB rather than disrupting the active gB-gH-gL complex. Among the 31 compounds analyzed, only 11 were found to be docking into gB active site, whereas 20 compounds were found to be docking into gH-gL active site, suggesting gH-gL complex active site as much preferred binding site for the ligand, in a given choice between both these glycoproteins active sites. |
| Â |
| Prediction of ADMET properties |
| Â |
| ADMET predictions are based on the molecular descriptor values (Table 4), according to Lipinski's rule of five, Veber's rule, and LAZAR online server. As the present work is concentrated only on the compounds which showed lesser binding energy than the CPMTHB, we will discuss about the ADMET predictions for these 31 compounds only. |
| Â |
|
|
Figure 7: a) Top view b) Front view of the grid box designed for the semi-flexible docking studies, so that only the active sites of gB and gH-gL glycoproteins should be searched for the ligand binding ability which were kept at ∼70 Å distance. |
|
| Â |
| Based on the experimental values, it was inferred that the tested compounds successfully satisfied all the parameters of Lipinski’s Rule of Five [24] i.e., the molecular weight must be |
| Â |
| The hydrophobicity of drugs could be inferred from Log P value. Log P Values are directly proportional to the oral hydrophobicity of the drug. Greater the hydrophobicity of the drug, higher will be its ability to circulate longer in our body. It wouldn’t be easy to excrete such a drug. In the present investigation, the Log P values of the drug molecules were observed to be in the range of 2.6 to 6.8. The highest Log P value of 6.8 was observed for compound ZINC40883455, which shows that it is the most hydrophobic molecule amongst all the analyzed. The least log P value amongst the analyzed compounds was compound ZINC66066505, which shows it as a most hydrophilic compound among the analyzed. However, all the compounds except compounds ZINC40883455, ZINC01936038, ZINC10154846, ZINC01941422, ZINC57470704, ZINC00515325 and ZINC07780416 were shown good hydrophobicity, with a range between 2-5 log P value. |
| Â |
| As per the Veber et al. [25], oral bioavailability of drugs could be measured by the molecular weight, number of rotatable bonds (n rotb), number of hydrogen bonds, and the expanse of the drug’s polar surface (TPSA). The oral bioavailability was marked by small molecular weight (less than 500); also, the number of rotatable bond must be less than 10, the number of hydrogen bond donors and acceptors must be less than 12, and TPSA values must be less than 140. Table 4 shows that all the compounds have a good oral bioavailability. |
| Â |
|
|
Table 4: The molecular descriptor values of the 31 best compounds along with their ZINC compound IDs. |
|
| Â |
| Toxicity predictions for the compounds analyzed was carried out using Lazar online server. Lazar is a software package which is used to detect mutagenic, and/or carcinogenic properties based on the similarities in functional group. Using Lazar, the toxicity and mutagenicity of compounds was verified by conducting an assay with Salmonella typhimurium. Table 5 shows that all the compounds analyzed do not have any mutagenic properties. On the other hand, the carcinogenicity of compounds was verified by animal testing studies with Mouse, shows that all the compounds analyzed have no carcinogenicity (Table 5). The descriptor values along with LAZAR results for all the 505 compounds analyzed in the present study can be found in supplementary material 2. |
| Â |
|
|
Table 5: Toxicity of compounds based on LAZAR results. |
|
| Â |
| Conclusions |
| Â |
| In this work, we discovered several compounds that are potentially able to block the interaction between active residues of gB and gHgL complex, suggesting their capability to inhibit the viral fusion and entry into the host cell. Compounds CPMTHB, ZINC01972391 and ZINC04799874 are found to be common strong inhibitors for both gB and gH-gL glycoproteins. From our structure based virtual screening studies coupled with semi-flexible and flexible docking studies, we propose that the tested compounds have the capability to attenuate the gB-gH-gL complex formation, rather than their capability to disrupt the already formed active complex. |
| Â |
| All the compounds we discovered in this work are found to bind either gB or gH-gL active binding sites, by creating hydrogen bonds and VDW interactions with important residues in the active sites. We All the compounds we discovered in this work are found to bind either gB or gH-gL active binding sites, by creating hydrogen bonds and VDW interactions with important residues in the active sites. We performed a detailed analysis of the atomic interactions between each potential compound, and residues inside the gB and gH-gL active sites to identify which residues interacted with the compounds. We have shown that the interaction between all compounds with the gB and gHgL active sites are facilitated by hydrogen bonds and VDW interactions, with atleast one of the active residues that are vital for gB-gH-gL complex formation. Moreover, the ADMET properties of these compounds are in accordance with Lipinski’s and Veber’s rules, which are most widely taken as the guidelines in designing drugs. Therefore, these compounds may be used as such or can be further optimized as leads for developing effective anti herpes drugs targeting this mode of action. |
| Â |
| This study is a step forward in elucidating gB-gH-gL complex interface as a potent anti herpes drug target. Our computational analysis provided a rationalization to the ability of the tested compound to attenuate this complex. The large value of binding energy involved in binding of these tested compounds consolidates the thermodynamic stability of the binding. Our docking results obtained substantiate the hypothesis that this tested compound has the potential to inhibit the association of gB to gH-gL, by inhibiting either of these glycoproteins, suggesting the possible mode of action of these tested compounds to inhibit herpes virus, by attenuating this complex formation, thus leaving significant evidence in support of this complex as a promising anti herpes drug target. |
| |
| Â |
| Methods |
| Â |
| Software and program |
| Â |
| Discovery studio Visualizer (Accelrys, Inc., USA) [26] is utilized to visualize the receptors, ligand structures, hydrogen bonding network, to calculate length of the bonds and to render images. The ZINC database [27] is a chemical compound database of commercially-available compounds, which contains over 21 million purchasable compounds in ready-to-dock, 3D formats, was used throughout this study to screen for potential inhibitors, based on structural similarities with the known gH-gL inhibitor CPMTHB, along with the molecular descriptor values. Arguslab 4.0.1 [28] was the primary docking program used in this work for the structure based virtual screening. AutoDock 4.0 [29] was used for performing semi-flexible and flexible docking studies, and the preparation of the ligands and protein receptors in pdbqt file and determination of the grid box size were carried out, using Auto- Dock Tools version 1.5.4 (The Scripps Research Institute, La Jolla, USA) [30]. All the AutoDock docking runs were performed in Pentium(R) Dual-Core CPU @ 3.00 GHz, with 2 GB DDR RAM. Auto-Dock 4.0 was compiled and run under Linux Ubuntu operating system. Lazar (https://lazar.in-silico.de/predict) online server was used for toxicology predictions. |
| Â |
| Preparation of gB and gH-gL structures |
| Â |
| The three-dimensional structures of gB [PDB:2GUM] and gHgL [PDB:3M1C] complex was retrieved from the Protein Data Bank. These structures were prepared by removing all bound crystal water molecules, ligands and hydrogen bonds were added. So obtained structures were saved in pdb files for further studies. |
| Â |
| Ligand-based virtual screening |
| Â |
| Virtual screening based on the structure of gH-gL inhibitor CPMTHB was carried out using the ZINC database. Two-dimensional structure of CPMTHB was used to search for similar compounds in the ZINC database, from the most common chemical suppliers (ChemBridge, ChemDiv, Ryan, Asinnex, MayBridge, Sigma-Aldrich, Comgenex, Otava and Specs), covering over 21 million chemical compounds. A total of 505 compounds with a threshold limit of 50% similarity, were identified to be structurally similar with CPMTHB. These 505 compounds were used in further molecular docking analysis. |
| Â |
| Structure-based virtual screening |
| Â |
| Arguslab 4.0.1 was utilized to search for potential inhibitors amongst the 505 compounds found through ligand based virtual screening, targeting gB and gH-gL active sites. We first carried virtual screen onto the gH-gL glycoprotein crystal structure, targeting its active residues, and then onto the active site of gB glycoprotein crystal structure, separately. Docking between receptor and ligands database in .sdf format was performed using “Dock a Database†option of Arguslab 4.0.1 software. A spacing of 0.4 Å between the grid points was used. Lamarckian Genetic Algorithm (LGA) [29] was selected as docking engine with the default parameters. "Dock" was chosen as the calculation type, “Flexible†for the ligand, and “AScore†was used as the scoring function. The grid box was set to 12, 23 and 21 Å (x, y, and z) cube for gB active site, and 16, 21 and 18 Å (x, y, and z) for gH-gL active site. |
| Â |
| Semi-flexible docking |
| Â |
| AutoDock 4.0 suite was used as molecular-docking tool, in order to carry out semi-flexible and flexible docking simulations. It is well established that AutoDock is reliable in locating docking modes that are consistent with X-ray crystal structures [31,32]. As described elsewhere in detail, Autodock assists in simulating interactions between receptors and drug compounds, allowing ligand flexibility [29]. Semi-flexible docking simulations were carried out by making CPMTHB and 31 better compounds flexible, while keeping the receptor macromolecules gB [PDB: 2GUM] and gH-gL glycoproteins [PDB: 3M1C] rigid. Flexibility of the ligand helps in exploring the spatial degrees of freedom for rotation and translation, for given number of torsional degrees of freedom. Interaction energy for every new location and conformation of the ligand is evaluated by applying a random perturbation for each time step [33]. The grid box was set to 126, 66 and 68 Ã… (x, y, and z), with 0.6 angstroms grid points spacing, by taking care that the grid box was designed, so that the active sites of both gB and gH-gL complex were surrounded by the three dimensional grid box. Lamarckian Genetic Algorithm (LGA) was selected as docking engine, with all the docking parameters set to default. After each LGA run, Autodock reports the best docking solution (lowest docked free energy), and results are reported based on cluster analysis. From a total of 10 docking modes represented by LGA cluster analysis, the lowest energy docking mode was selected from each docking simulation. |
| Â |
| Flexible docking |
| Â |
| AutoDock 4.0 has a novel feature for allowing the side chains in the protein, as well as in the ligand to be flexible, which can be achieved by making use of AutoDock flexres scripts. As there is every possibility that the ligand will try to make its own interactions with the residues in the active sites of the two glycoproteins, in order to minimize the energy, we made the key residues of gB and gH-gL glycoproteins, which were found to be forming H-bonds with corresponding residues in the two glycoproteins, as reported [18] flexible, for observing the mode of interactions of these key residues with the incoming ligand. AutoDock Tools, a Graphical User Interface program was utilized in preparing the protein receptors, ligand molecules, and to analyze the docking simulations using default protocols [34]. A range of 4-9 bonds in the ligands were made “active†or “rotatableâ€. Atomic salvation parameters were assigned to the receptor using default parameters. The location and dimensions of the grid box are chosen, such that it incorporates the key residues which are involved in gB-gH-gL active complex formation. The energy scoring grid was prepared as a 60, 40 and 86 Ã… (x, y, and z), with 0.375 angstroms grid spacing points. Lamarckian Genetic Algorithm was selected as docking engine with all the docking parameters set to default. Gibbs free energy (ΔG) is calculated as a sum of six energy terms of dispersion/repulsion, hydrogen bonding, electrostatic interactions, deviation from covalent geometry, internal ligand torsional constraints, and desolvation effects. |
| Â |
| Acknowledgement |
| Â |
| The authors wish to acknowledge the assistance of T. Deepthi, in acquisition of Lazar data. Syed Hussain Basha specially thank and dedicate this work to his parents Syed Abdul Khader Basha and Syed Khursheed Begum for their constant encouragement and much needed support, without which this research work wouldn’t have been possible. |
| Â |
| Authors’ contribution |
| Â |
| Syed Hussain Basha conceived and designed the experiments. Syed Hussain Basha and K Naresh Kumar performed the experiments. Syed Hussain Basha analyzed the data. Syed Hussain Basha wrote the paper. K Naresh Kumar helped with editing the paper. Both the authors have read and approved the manuscript. |
| Â |
| |
| References |
| Â |
- Xu F, Sternberg MR, Kottiri BJ, McQuillan GM, Lee FK, et al. (2006) Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 296: 964-973.
- Cusini M, Ghislanzoni M (2001) The importance of diagnosing genital herpes. J Antimicrob Chemother 47: 9-16.
- Paz-Bailey G, Ramaswamy M, Hawkes SJ, Geretti AM (2007) Herpes simplex virus type 2: epidemiology and management options in developing countries. Sex Transm Infect 83: 16-22.
- Gupta R, Warren T, Wald A (2007) Genital herpes. Lancet 22: 2127-2137.
- Smith PD, Roberts CM (2009) American College Health Association annual Pap test and sexually transmitted infection survey: 2006. J Am Coll Health 57: 389-394.
- Roberts CM, Pfister JR, Spear SJ (2003) Increasing proportion of herpes simplex virus type 1 as a cause of genital herpes infection in college students. Sex Transm Dis 30: 797-800.
- Looker KJ, Garnett GP, Schmid GP (2008) An estimate of the global prevalence and incidence of herpes simplex virus type 2 infection. World Health Organization 86: 737-816.
- Rudnick CM, Hoekzema GS (2002) Neonatal herpes simplex virus infections. Am Fam Physician 65: 1138-1142.
- Beyrer C, Jitwatcharanan K, Natpratan C, Kaewvichit R, Nelson KE, et al. (1998) Molecular methods for the diagnosis of genital ulcer disease in a sexually transmitted disease clinic population in northern Thailand: predominance of herpes simplex virus infection. J Infect Dis 178: 243-246.
- Chen CY, Ballard RC, Beck-Sague CM, Dangor Y, Radebe F, et al. (2000) Human immunodeficiency virus infection and genital ulcer disease in South Africa: the herpetic connection. Sex Transm Dis 27: 21-29.
- Mwansasu A, Mwakagile D, Haarr L, Langeland N (2002) Detection of HSV-2 in genital ulcers from STD patients in Dar es Salaam, Tanzania. J Clin Virol 24: 183-192.
- Wald A, Link K (2002) Risk of human immunodeficiency virus infection in herpes simplex virus type 2-seropositive persons: a meta-analysis. J Infect Dis 185: 45-52.
- Fleming DT, McQuillan GM, Johnson RE, Nahmias AJ, Aral SO, et al. (1997) Herpes simplex virus type 2 in the United States, 1976 to 1994. N Engl J Med 337: 1105-1111.
- Armstrong GL, Schillinger J, Markowitz L, Nahmias AJ, Johnson RE, et al. (2001) Incidence of herpes simplex virus type 2 infection in the United States. Am J Epidemiol 153: 912-920.
- Spear PG, Longnecker R (2003) Herpesvirus entry: an update. J Virol 77: 10179-10185.
- Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, et al. (2010) Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat Struct Mol Biol 17: 882-888.
- Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, et al. (2006) Crystal structure of glycoprotein B from Herpes simplex virus 1. Science 313: 217-220.
- Syed HB, Deepthi T, Nalini RP (2013) Computational repositioning of ethno medicine elucidated gB-gH-gL complex as novel anti herpes drug target. BMC Complementary and Alternative Medicine 13: 85.
- Peng T, Ponce de Leon M, Novotny MJ, Jiang H, Lambris JD, et al. (1998) Structural and antigenic analysis of a truncated form of the herpes simplex virus glycoprotein gH-gL complex. J Virol 72: 6092-6103.
- Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, et al. (2010) Bimolecular complementation defines functional regions of Herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J Virol 84: 3825-3834.
- Whitley RJ, Gnann JW Jr (1992) Acyclovir: a decade later. N Engl J Med 327: 782-789.
- Hirsch MS, Swartz MN (1980) Drug therapy: antiviral agents (second of two parts). N Engl J Med 302: 949-953.
- Chatis PA, Crumpacker CS (1992) Resistance of herpesviruses to antiviral drugs. Antimicrob Agents Chemother 36: 1589-1595.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23: 3-25.
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, et al. (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45: 2615-2623.
- Accelrys Software Inc. (2011) Discovery studio Visualizer 3.0
- Irwin JJ, Shoichet BK (2005) ZINC–A free database of commercially available compounds for virtual screening. J Chem Inf Model 45: 177-182.
- Mark AT, ArgusLab 4.0.1, Planaria Software LLC, Seattle.
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, et al. (1998) Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem 19: 1639-1662.
- Sanner MF (1999) Python: a programming language for software integration and development. J Mol Graph Model 17: 57-61.
- Dym O, Xenarios I, Ke H, Colicelli J (2002) Molecular docking of competitive phosphodiesterase inhibitors. Molecular Pharmacol 61: 20-25.
- Rao MS, Olson AJ (1999) Modelling of factor Xa-inhibitor complexes: a computational flexible docking approach. Proteins 34: 173-183.
- Goodsell DS, Morris GM, Olson AJ (1996) Automated docking of flexible ligands. Applications of AutoDock. J Mol Recognit 9: 1-5.
- https://www.scribd.com/doc/61267608/UsingAutoDock4forVirtualScreening-v4.
|
| Â |
| Â |