Review Article Open Access
Alzheimer's Disease Pathogenesis: The Denied Access Model
Wade ND and Shaohua X*Department of Biology, Florida Institute of Technology 150 W. University Blvd, Melbourne, Australia
- Corresponding Author:
- Shaohua Xu
Department of Biology
Florida Institute of Technology 150 W. University Blvd
Melbourne, Australia
Tel: (321) 674-8430
E-mail: xshaohua@fit.edu
Received date: June 16, 2017; Accepted date: July 12, 2017; Published date: July 18, 2017
Citation: Wade ND, Shaohua X (2017) On Alzheimer’s Disease Pathogenesis: The Denied Access Model. J Alzheimers Dis Parkinsonism 7:359. doi:10.4172/2161-0460.1000359
Copyright: © 2017 Wade ND, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Alzheimers Disease & Parkinsonism
Abstract
Currently, AD has no cure and only treatments for the symptoms exist. Modern research still debates the toxic component of AD and the exact mechanism causing neurodegeneration. A hallmark of the disease is the production of the amyloid-beta (Aß) peptides and eventual self-assembly of these peptides into fibrils and extracellular Aß plaques. Both plaques and oligomers are proposed to be the direct cause of AD, but it remains unclear how the physical presence of these structures affect neuronal function and pathogenesis. Biomolecule aggregation is known to play a role in the pathogenesis of numerous diseases by restricting diffusion and bulk flow, and the same restriction could occur in the brain due to the dense amyloid plaques forming in the extracellular space. These plaques could prevent proper flow and diffusion of essential nutrients and prevent cellular waste removal by acting as extracellular channel blockades; however, limited models exist that address these issues. Alternative models and molecular tools need to be developed which focus on diffusion and bulk flow in relation to neural function and the physical presence of amyloid plaques. This review aims to evaluate the effect of the plaques on diffusion and bulk flow in relation to neural function in the brain.
Keywords
Neurodegeneration; Alzheimer’s disease; Pathogenesis; Plaques; Peptide fragment
Introduction
Obstruction of bulk flow by aggregated biomolecules is thought to play an important role in the pathogenesis of numerous human diseases, such as kidney stones, gallbladder stones, atherosclerosis, and thrombotic cardiovascular diseases [1]. Amyloid plaques deposited in the human brain are derived from amyloid-beta (Aß) peptides that aggregate and form fibrils which bundle to form plaques [2]. These plaques are also made of other proteins, lipids, and their calcification. How the presence of plaques affects neuronal function and pathogenesis, and whether the amyloid plaques obstruct flow of fluid molecules is unclear. Compared to the centimeter large atherosclerotic plaques or kidney stones, amyloid plaques are smaller, often less than 50 μm in diameter, making them a difficult subject to isolate and study.
The AD field has shifted from studying the potential toxicity of the plaques to studying the toxicity of the intermediate oligomers [3]. Some groups believe the plaques represent a non-toxic byproduct and provide a neuroprotective role by sequestering the oligomers [4-7]. After years of oligomer-focused research, numerous inconsistent results and lack of evident in vivo toxicity exists [8]. New methods are needed to study the potential physical toxicity of the plaques in relation to diffusion and bulk flow and to study the physical relationship of extracellular protein aggregation to viscosity and neuronal function. The cause of the massive amount of cell death found in AD remains unexplained so the search for an alternative toxicity is needed.
Aggregation and transformation of misfolded proteins into amyloid fibrils is a commonly observed phenomenon in dozens of human diseases, ranging from diabetes mellitus type 2 to neurodegenerative diseases such as Alzheimer’s disease (AD), Huntington’s disease, and Parkinson’s disease [9]. Since the human body has no known mechanism for removing these amyloids, patients tend to already have extensive amyloid deposits once diagnosed, which leads to very poor prognoses [10]. Specifically in AD, Aβ accumulation with time leads to the formation of amyloid plaques [11].
Currently, the pathogenesis of Alzheimer’s disease remains unknown. Two leading hypotheses have been proposed: 1) amyloid plaques are pathogenic, and more recently, 2) the oligomer intermediates or as we refer to them, colloids, of fibril formation is pathogenic. Amyloid fibrils can further aggregate into a gel and there is a potential link between gelation and cellular death, as gels are known to eliminate bulk flow at the macroscopic level [12]. However, bulk flow at the microscopic level, under hydrostatic pressure, remains an area of study. New methods need to be developed to determine if amyloid plaques obstruct fluid flow and the circulation of nutrients, waste, and bio-signaling molecules through brain tissues and extracellular space.
Materials and Methods
Ad pathogenesis: Oligomer vs. amyloid hypotheses
Aggregation and accumulation of two different protein molecules causes pathogenesis in AD. Tau, a protein associated with cytoskeleton assembly, accumulates intracellularly to form macromolecule networks called neurofibrillary tangles (NFTs). Aβ, a peptide fragment, accumulates in the extracellular space in structures called plaques. Virtually all neurons that degenerate during the development of AD have accumulation of one or both forms of aggregation; however, Aβ formation alone can attribute to the disease [13].
Amyloid beta oligomers and aggregation
Once the Aß40 or Aß42 peptide is produced, the exact pathways and mechanisms of amyloid aggregation at the molecular level are poorly understood, which hinders rational pharmaceutical interventions. Critical intermediates in this pathway, colloids and their linear aggregates, composed of highly ordered β-sheet complexes [14] were discovered using atomic force microscopy [15] and later confirmed using transmission electron microscopy [16].
Protein molecules initially aggregate into colloidal spheres of uniform size, which then join together linearly, forming amyloid fibrils and eventual plaques (Figure 1) [17,18]. These insoluble plaques are commonly associated with cell death [3] and may lead to progressive degeneration of the blood-brain barrier (BBB) [19]. The accumulation of Aβ plaques in the extracellular space of the brain may initiate a cascade of reactions, beginning with synaptic dysfunction and ultimately resulting in apoptosis of the neuron [3,20-22]. The plaques may form around axons [23] and become associated with activated microglia and reactive astrocytes [11], which produce toxic molecules, such as cytokines, causing dysfunction of neuronal processes.

Figure 1: Proposed mechanism of colloidal aggregation leading to amyloid fibril formation [17].
Cytokines increase the inflammation of the neuron [24] and increase the production of toxic reactive oxygen species [11].
Currently, the popular amyloid cascade hypothesis (ACH), first proposed by Hardy and Higgins [25], has come under question due to formation of plaques in individuals who have normal cognitive function [26,27]. Alternatively, the oligomer hypothesis suggests the colloidal intermediates, also known as oligomers, are the toxic component that leads to neurodegeneration [28,29]. Aß Oligomers (AßOs) are either a step in the formation process or a byproduct of Aβ plaques [30]. The validity of the oligomer hypothesis depends on whether oligomers were present earlier than the fibrils and plaques and exercise their toxicity before substantial plaque formation.
As for the ACH, plaque formation may not have followed the proposed nucleation pathway an oligomers may be a byproduct or produced by other mechanisms [30].
Interestingly, a mouse model supporting the oligomer hypothesis displayed cognitive decline before substantial plaque formation [31]; however, supporting in vivo work at normal concentrations is lacking. The possibility also exists where the plaques provide a neuroprotective role [32] or are simply an innocent bystander in cell death.
The main issue with the oligomer hypothesis is the diversity of toxicity (Figure 2). Countless articles are published which present new mechanistic hypotheses of AßO toxicity. To briefly summarize, AßOs have been linked to AD in the following ways: Oxidative stress and lipid peroxidation in neurons [33-36] toxic ion pore formation on the cell membrane [37,38], synaptic plasticity disruption [39- 42], astrocyte and microglia calcium influx [36], selective neuronal degeneration [43], prion-like infectious behavior [34,44,45], binding to numerous membrane proteins [46-49], insulin resistance [50-52] calcium homeostasis disruption [35,53] and tau hyperphosphorylation induction [54]. One of the main issues is that AßOs lack a common description of structural toxicity and are thought of as “an emperor in need of clothes” since they possess numerous conformations ranging from monomers, to trimers and to eventual fibrils [8]. Moreover, not all oligomers promote toxicity [54,55] and “relevant Aß toxicity has barely been demonstrated” [56].

Figure 2: The diverse oligomer theory [48].
If AßOs are the toxic component, the massive cell loss, role in cognitive alterations and existence in non-AD patients cannot be fully explained [8]. More in vivo work using naturally occurring concentrations of AßOs along with other models of toxicity must be examined. Also, if they are toxic at low concentrations, such as 100 nM, why is toxicity specific to neurons and not elsewhere in the body where AßOs are also found?
The failure of many clinical trials following the ACH has caused researchers to change focus towards the oligomer theory. Most of the failures are due to the pitfalls of the transgenic mouse AD models and their inability to successfully correlate clinical trials to the complex human system [57,58]. Interestingly, the only FDA approved clinical drugs were never tested in the transgenic mouse model [57].
The ACH is also very unappealing for therapeutic treatment [59], since even if clearance of the plaques is successful, the surrounding neurons are already dead or damaged, so cognition cannot be restored. From a clinical view, attacking the oligomers appears much more appealing, and has made them the key drug target [60] and the focus of recent proposals. Clinical trials targeting oligomers have also failed across the board, again likely linked to the imperfect mouse model; however, trials have not focused on all aspects of the disease. Future trials must focus on all aspects including oligomers, fibrils, plaques and even tau [61]. Although some groups believe it, the plaques should not be considered a non-toxic byproduct, and should remain as a research focus based on their potential physical toxicity. Since existing models have not been successful, new models and methods must be proposed to address the issue of plaque’s physical presence. Much debate remains as to which aggregation state is toxic to the cells [62,63] and more work elucidating the physical effects of plaques within the brain must be investigated. It is likely the two disease states are linked, and both play a role in toxicity.
Results
Why denied access model?
In vitro, amyloid fibers have been found to be capable of further aggregating and forming gels [12,64]. Gels are known to be capable of eliminating bulk flow and reduce diffusion. Can amyloid plaques in AD be gels of porous matrix? TEM images of amyloid plaques either in AD tissue or isolated by density gradient centrifugation reveal these plaques are biogels with dense fiber network [64,65]. A transformation of brain fluid to a gel-like state due to amyloid fibril formation could have drastic neurophysiological effects on viscosity-dependent processes and tortuosity, or obstructive geometry, of the extracellular space.
Denied access model
When surrounded by amyloid plaques/protein gel, neurites are denied to have free access to interstitial ions, nutrients, signaling molecules and metabolic waste drainage pathways, which results in the inhibition of the compound action potential propagation. This is an alternative model for the AD community to consider, although continued investigations are required to verify the model and to fulfill the details. We hypothesize that gel formation around axons may inhibit the heart-pulse-driven fluid flow and then the circulation of nutrients and ions around the axon and affect the propagation of the action potential.
Denied access model explains numerous observations associated with ad tissue analysis
In Alzheimer’s disease, extensive neuronal cell death is found in anatomical regions of the brain where there is no presence or accumulation of neuritic senile plaques, and/or paired-helical filaments (PHF). Based on our model, plaques wrapped around axons may affect the vitality of the neuronal cell bodies located in a different anatomical region. Cognitive impairment and the degree of dementia do not correlate with the amyloid-beta plaques burden in many Alzheimer’s disease patients. However, the amyloid-beta plaque burden is quantified not based on the type of the plaque but rather a sum of all types. Based on our model, primitive plaques may not change the bulk flow or viscosity as much as the classical plaque. Thus, there exists a possibility that the classical plaque burden correlates better with the stages of Alzheimer’s disease. Our model also explains the observation that in transgenic mice amyloid plaques, although not classical ones, are found, but neuronal and synapse loss are rather minimal.
The denied access model also applies to other diseases where accumulation of protein aggregates may affect local diffusion, bulk flow and bioenergy-driven transportation. Since the toxic effect is physical rather than chemical, the model applies to not only AD but also alphasynuclein aggregation-induced Parkinson’s disease, aggregation of prion protein induced transmissible spongiform encephalopathies and islet amyloid polypeptide aggregation induced death of the beta cell in type II diabetes. Movement of molecules, ions and subcellular particles will all be affected due to the aggregation of proteins and then gelation of the amyloid fibers.
The denied access model is simple and inclusive of the physics and engineering perspective of gels or plaques, in addition to the biochemical nature. The inclusion of physical properties such as bulk flow and diffusion is seen in the illustration of atherosclerosis, thrombosis, gallbladder stones and kidney stones, where aggregation of biomolecules, large and small, is also an early event of disease pathogenesis. The transportation phenomenon, or bulk flow and diffusion, through gels are an active research area in engineering.
Denied Access by Attenuated Diffusion and Bulk Flow Through the Amyliod Plaque
The extracellular space (ECS) defines the channel-like environment where interstitial fluids (ISF), consisting of nutrients, neurotransmitters, signaling molecules, ions, and wastes, travel through the all tissues to maintain cellular homeostasis and function. The widths of the channels range from 20-40 nm and encompass around 20% of total brain volume [66,67]. Variations of the ECS geometry and volume fraction can arise during sleep, development, aging and neurological disorders, so understanding any alterations in function are vital for neurological research and drug delivery. These variations can affect proper clearance routes, cellular uptake, diffusion, and flow of nutrients and waste across the BBB [68].
Diffusion
Diffusion is the net movement of individual molecules down a concentration gradient due to random motion or thermal movement. Diffusion was first defined by Adolf Fick (Eq. 1) in the 19th century as,
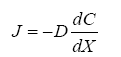 (1)
(1)
Where J is the flux through an area, D is the diffusion coefficient, dC is the change in concentration of material, and dx is the change in distance. Thus, flux is proportional to the concentration gradient. Diffusion through the ECS is determined by the volume fraction or porosity, defined as α (Eq. 2) and the tortuosity or obstructive geometric capabilities of the environment, defined as λ (Eq. 3) HYPERLINK\l “page36” [68]. Volume fraction is defined as,
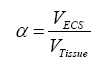 (2)
(2)
where VECS is the volume of the extracellular space devoid of matrix components and VTissue is the volume occupied by the tissue, and tortuosity is defined as,
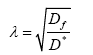 (3)
(3)
where Df is the free diffusion coefficient and D* is the effective diffusion coefficient, typically determined using radiotracers or fluorescent dyes. The volume fraction of the ECS has been well studied using electron microscopy and occupies approximately 20% of total brain volume [69,70]. This value has been recently debated due to the difficulty in determining the ECS volume fraction in living tissues, where there is a high likelihood of ischemia; therefore, new methods, such as in vivo studies using quantum dots [71] and fluorescent dyes [72], have been developed and remain an area of study.
Due to the physiological independence between volume fraction and tortuosity, it is difficult to derive a relationship between the two. There are also many non-geometric factors such as ISF viscosity (discussed in bulk flow), molecular size, ECS channel interaction [73] and the uptake of molecules by cells or across the BBB [74]. The Nicholson group attempted to derive an empirical relationship using Archie’s law (Eq.4) HYPERLINK \l “page27” [75-77],
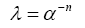 (4)
(4)
Which relates sedimentary porosity and saturation. It was determined that the values remained independent since the tortuosity exponent, n, is a variable according to the original law. However, if the tortuosity exponent is variable due to the variance between regions of white and gray matter, the relationship displays some dependence [78,79]. This relationship remains an important area of investigation when discussing diffusion throughout the brain.
Bulk flow
Bulk flow is the movement of water and solutes in response to a pressure gradient. Analysis of fluid flow through a porous matrix can be defined using a simplified version of
Darcy’s law (Eq. 5) which states,
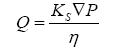 (5)
(5)
Where Q is the flux of fluid per unit area through a porous matrix, Ks is the Darcy permeability coefficient, ∇P is the hydrostatic pressure gradient across the matrix and η is the viscosity of the ISF. According to the equation, bulk flow is strongly dependent on the viscosity of the media and the permeability through a porous matrix. The movement of fluids through the ECS of the brain has numerous implications, including cell to cell communication, drug delivery, distribution, waste clearance, ionic homeostasis, and immune function [80].

Figure 3: Denied access model. The neurite regions surrounded by amyloid gel plaques are denied from accessing to nutrient and signaling molecules as well as to the drainage pathways for metabolic wastes. This denied access leads to the inhibition of the action potential propagation and eventually the degeneration of the disused neurites.
Bulk flow through the ECS remains a debated issue [81]. Flow typically occurs via osmotically balanced fluid secretion across the BBB but can also be generated as fluid secretion from cells [82] and the choroid plexuses [83]. One research group determined bulk flow only occurs in perivascular spaces surrounding capillaries and little to no flow occurs throughout the remaining ECS [84]. Varying flow rates were also found when comparing white to gray matter regions of the brain. White matter regions expressed a flow rate of 10.5 μm/min towards the ventricles, but regions of gray matter only displayed flow under osmotic stress [85]. The issue of diffusion also returns, since some groups believe that diffusion cannot be the sole clearance mechanism due to a constant rate of clearance for various sized molecules [86-88]. However, another group that used albumin and large dextrans suggests diffusion is responsible for clearance and is independent from bulk flow [89]. As mentioned, quantitative measurements of flow rates have been made by injecting tracers into the brain and observing their movements [85,90], but no work has examined direct effects of Aß plaques.
Limited research has been conducted towards understanding how the formation of amyloid plaques affects diffusion. Researchers utilizing diffusion-weighted magnetic resonance imaging (DW-MRI) have demonstrated a reduced interstitial fluid diffusion in the brain of APP23 transgenic mice [91] and reduced diffusivity in the white matter of patients suffering from sporadic Jakob-Creutzfeldt disease [92], another neuroencephalopathy associated with the formation of insoluble amyloid. Sykova [93] analyzed the ECS and the volume fraction and diffusion of the cerebral cortex in APP23 mice using realtime tetramethylammonium (TMA) and DW-MRI methods. They determined that the volume fraction increased in aged APP23 mice by approximately 3.0% in both female and male mice and apparent diffusion of TMA decreased by 7.0% in females and 1.7% in males. Plaque load was also twice as high in females as it was in males. The control group decreased their ECS volume fraction by approximately 7% in males and approximately 4% in females, and displayed a<1% increased apparent TMA diffusion in aged mice [93]. These results suggest that plaques may decrease diffusion by increasing overall brain tortuosity; however, the direct correlation remains unknown.
Another physical impact of plaque formations may be fluid clearance from the brain by increasing the tortuosity, as previously noted by Sykova et al. [93]. Clearance is important across the BBB to prevent buildup of toxic molecules and to help maintain cellular homeostasis (Figure 4A). Clearance also prevents high concentrations of soluble Aβ and eventual plaque formation [94,95] and an age-related imbalance of Aß release could drive the progression of AD [3]. The accumulation of plaques in the extracellular space (Figure 4B) and drainage pathways (Figures 4C and 4D) could reduce the clearance of high molecular weight substances by impeding the normal clearance routes [68,70,96,]. Plaques can become very large, up to 150 μm in diameter [97], so it is likely that the dense plaques can restrict fluid movement through the less than 0.1 μm extracellular space pathways. Moreover, AD patients display a significant reduction in ISF flow [98], which may be linked to the increased plaque load. Future work must develop new methods to evaluate the effect of the presence of amyloid plaques on neuronal function and fluid flow in brain tissue.

Figure 4: Bulk flow and fluid clearance in the brain. (A) Nutrients and toxins must be able to freely move into and out of the brain (Image from the University of New Hampshire). (B) Plaque formations can become very large, up to 150 μm in diameter [97] and can likely block the (C) less than 0.1 μm extracellular channels and synaptic junctions [70]. (D) Factors within the extracellular space such as charged residues, binding, and dead space can also affect fluid flow [68].
Discussion
Techniques to study amyloid plaques on neuronal function
We identified four new methods being developed in our laboratory to study if and how the amyloid plaque affects the neuronal function. Specifically, these methods will be used to analyze the following:
Fibrin as an aß plaque proxy
To study the effect of amyloid plaque on neuronal function, we need to have the plaque prepared in the presence of the functional neurons, under physiological conditions and within seconds to minutes. The constraints of the physiological condition and the short time period force us to look for a proxy of amyloid fibril gel as they take hours to days to produce, often under non-physiological conditions.
Volume fraction and pore size: Ideally, the use of Aβ is preferred, but time, attainability, and practicality must be considered. Fortunately, amyloid fibrils are not the only example of a fibrous network created by proteins. The hindrance of diffusion and convective transport are also elementary properties of wound healing, a process that relies on blood clotting due to the formation of a fibrin gel network. Fibrin gels are formed within a few minutes between the interactions of the protein fibrinogen, from blood plasma and the activating enzyme thrombin [99]. Thrombin cleaves fibrinopeptides located in the middle of fibrinogen, producing a fibrin monomer and subsequent protofibril formation [100]. Compared to amyloid gels, fibrin is easily attainable, found within the body, and forms rapidly under physiological conditions [99]. Amyloid gels, on the other hand, require hours or even days to form in vitro. Importantly, fibrin is physically similar to amyloid gels and provides an ideal proxy. With these factors considered, a convenient analog to study the physical effects of toxicity must be used. This would allow for rapid and feasible studies of protein aggregation in cellular environments.
Fibrin provides an ideal analog for studying the physical effects of amyloid plaques or gels and the potentially abolished bulk flow on neuronal function; however, the gel must have the appropriate porosity to simulate the amyloid plaque. To qualify, fibrin gel must have higher porosity compatible to amyloid gel. Scanning electron microscopy (SEM) (Figure 5A) and atomic force microscopy (AFM) (Figure 5B) images reveal a dense fibrous network with pore sizes less than 1 μm, which are comparable to Aβ (Figure 5C).

Figure 5: Scanning electron micrograph of fibrin (A), atomic force micrograph of fibrin (B), and atomic force micrograph of amyloid-beta (C). Fibrin was generated using a 10:1 ratio of fibrinogen (20 mg/ml in 0.9% NaCl) and thrombin (50 U/ml in 0.1% BSA, pH 7.4). Amyloid fibril formation of 50 uM Aβ42 fibrils incubated at 37°C is seen in 1 week [128].
Chemically, fibrin is different from Aβ, and although fibrin is not a true amyloid, its regulation is similar to the enzymatic control of peptide hormone aggregation [101]. Fibrin has a high β-sheet content of 40% [102] and can stain using Congo red [103], an amyloid-specific stain. Kranenburg [103] believe fibrin can form cross-β structures and amyloid fibrils, and that these β-sheet structures function as the scaffold to activate plasmin, which eventually cleaves aggregated fibrin.
Viscosity of gels using molecular rotors: Increased viscosity is also thought to play a role in the pathogeneses of AD linked to amyloid plaque formation. De la Torre and Mussivand [19] hypothesized that blood viscosity can increase due to amyloid deposits causing degeneration of brain capillaries at the BBB; however, the link between increased local viscosity and degeneration has remained elusive. Protein aggregation into a gel, as seen in vitro, possibly also occurs in vivo.
Bulk viscosity is known to be different from molecular viscosity. For fibrin to be qualified as an amyloid fibril gel proxy for diffusivity study, the viscosity of fibrin gel, at the molecular level, needs to be comparable or lower than that of amyloid fibril gel. To study ion or molecule diffusion across the amyloid plaque, the molecular viscosity is of interest. A successful technique used to study viscosity applies fluorescent molecules known as molecular rotors. These rotors are able to alter their fluorescence by changing the rate of an aromatic conformational change [104,105]. This rate of change is dependent on the microviscosity of the environment decreasing bond rotation and increasing fluorescent yield [106]. Previous work in the lab used 9-(2-carboxy-2-cyanovinyl) julolidine (CCVJ) as the molecular rotor to determine how the formation of fibrin altered environmental viscosity [107]. Results were compared to varying concentrations of glycerol, a known viscous solution, where increasing concentrations amplified the fluorescence intensity. In 20 mg/ml fibrin, fluorescence intensity increased during the incubation period with the greatest increase occurring within the first 60 s.
Only a small increase occurred in 10 mg/ml fibrin (Figure 6), suggesting 20 mg/ml would be an optimal concentration to study gelation in a time dependent manner. This work established that gelation results in an increased environmental microviscosity.

Figure 6: Relative fluorescence intensity of 5 μM CCVJ in two different fibrin gels. One gel was composed of 20 mg/mL fibrinogen (�?�), and the other was composed of 10 mg/mL fibrinogen (�?�). Thrombin concentration was kept constant (10-75 U/mL).
Sciatic nerve as a CNS proxy
A goal is to determine whether the physical presence of plaques causes toxicity. Since in vivo experiments using mice or human models are difficult to perform, an ideal analog is the sciatic nerve from frogs, such as the Xenopus laevis, as it has long been successfully employed in studying numerous physiological properties of axons and is well characterized. The sciatic nerve is a large bundle of many axons surrounded by an epineurium membrane (Figure 7A). The individual axons are structurally supported with collagen, known as the endoneurium, and encompassed with myelin containing nodes of Ranvier (Figures 7B and 7C), which allows for rapid conduction of action potentials.

Figure 7: Scanning electron micrograph images of Xenopus laevis sciatic nerve. (A) The sciatic nerve image was generated by fixing the nerve bundle in 3% glutaraldehyde, washing in 0.1% PBS, staining in 1% osmium tetroxide/0.1% PBS, dehydrating in increasing concentrations of ethanol and ethyl-acetate, and critical point drying. Cryostat sections were also prepared in the cross sectional (C) and longitudinal (D) directions at 20 μm thickness.
In AD, white matter atrophy due to plaque formation may be as important as gray matter atrophy. Plaques are more numerous in associative regions of the brain higher in gray matter content, such as the extracellular matrix of the hippocampus where learning and memory take place [108] and where the most severe neuropathological changes occur [22]; however, plaques are also found in myelinated white matter regions such as the cortex. The question arises as to whether the myelinated sciatic nerve would serve as an appropriate proxy for neurodegenerative studies. Although the gray matter regions are more intensely studied, white matter atrophy occurs in AD [109] and is found in roughly 60% of AD cases [110]. White matter atrophy is often considered secondary to gray matter damage [111], but recent developments determined white matter damage occurs in pre-AD stages and cannot be linked to gray matter atrophy [112,113]. Furthermore, studies analyzing white matter attributed the damage to cognitive decline [114] and discovered higher levels of soluble Aβ in AD patients [115].
Many physiological experiments using the frog sciatic nerve have been conducted examining the compound action potential (CAP) and the biochemical effects that may cause degeneration. One group examined the time-dependent effects acrylamide had on the antioxidative stress in rat nerve tissues. With increased concentrations injected into rats over a few weeks, decreases were found in both CAP duration and CAP amplitude [116].
Furthermore, [116] determined that acrylamide may be associated with an increase in lipid peroxidation and reduction of the antioxidative capacity. Another experiment examined the direct effects of polyethylene glycol (PEG) on isolated rabbit sciatic nerves [117]. They found 40% PEG abolished the compound action potential within an hour due to increased osmotic pressures creating a hypertonic environment and shrinkage of the axon. These structural changes may lead to reduced ion permeability, neuronal stress, and eventually death [117]. These results suggest the frog sciatic nerve may be an ideal tissue for neurophysiology experimentation to test the effect of viscosity on neuronal function. Completing these experiments on gray matter would be extremely difficult; whereas, the sciatic nerve is easy to dissect and shares common physiological features as central nervous system neurons [118]. If plaques are the culprit for neuronal loss, similar reasons for toxicity may be seen in both white and gray matter.
Quantifying diffusion through plaques and brain tissue
How amyloid plaques affect the circulation of nutrients, wastes and biosignaling molecules in brain tissue remains unknown. Molecules move through a matrix either by diffusion driven by Brownian motion, or carried by fluid movement or bulk flow. Thus, methods are needed to analyze diffusion and bulk flow. FRAP is a straightforward technique developed to study molecular diffusion in media [119]. Using a confocal laser scanning microscope (CLSM), a user-defined region of interest is selected and bleached, leaving the surrounding area unbleached [120].
With time, the unbleached molecules diffuse into the bleach area and exchange with the bleached molecules [121,122]. Using one of many possible fluorescent dyes, such as rhodamine B (RhoB) or fluorescently tagged proteins, a fluorescent recovery curve can be generated and a diffusion coefficient of the dye through the sample can be calculated [123,124].
Protocols and data analysis of FRAP experiments differ between laboratories causing the existing body of data to be incomparable [125]. Many finite factors affect diffusion, suggesting that each lab must perfect their own calibrations and controls. Numerous studies using this technique and a variety of fluorophores have been completed; however, one vital element of experimental approach arises, fixation of the tissue. Most FRAP experiments analyze real-time protein movement and interactions requiring live tissue samples and cell cultures. If the methods advance for fixed tissue use, countless studies could be completed and experimental procedures would be much simpler.
Effect of plaques on bulk flow
The physical presence of amyloid plaques may hinder brain clearance and deter the movement of nutrients and wastes in the brain. At the tissue level, experiments injecting molecular tracers [85,90] and studies using positron emission tomography [98] have evaluated bulk flow, but they are difficult to perform for standard laboratories. These experiments were focused on movement into and out of the brain rather than through specific regions. Other experiments also focused on either CSF or ISF production rates in relation to bulk flow [126] or used computer based modeling to analyze flow [127]. Feasible, reliable, and straightforward techniques are needed to study fluid movement at the cellular level and to study the relationship of flow due to individual plaques.
It is possible to estimate the bulk flow if the diffusion coefficient of a particular molecule is known through a plaque and free medium. Knowing these will allow the tortuosity and void volume fraction of the plaque to be estimated. That information can then be used to estimate Darcy’s permeability through the plaque and the effect on bulk flow compared to normal flow through the ECS.
Conclusion
Gelation’s relationship to increased viscosity and reduction in bulk flow is a common phenomenon in physical chemistry and engineering, but the increased viscosity has never been confirmed in Aβ plaques found in Alzheimer’s disease (AD). In vitro, proteins, such as Aβ and lysozyme, can undergo conformational changes and self-assemble into amyloid fibrils.
Eventually these fibrils can aggregate and form highly viscous, rigid gels [12,63]. In vivo, it is likely that amyloid plaques act as viscous and structured gels. The elimination of bulk flow and reduced diffusivity due plaque formation in the ECS near axons may be responsible for the inhibition of action potential propagation, which, in time, may cause neuronal death. Here we introduce the denied access model: when surrounded by amyloid gel plaques, neurites are denied access to nutrients, signaling molecules, and metabolic waste drainage pathways, eventually leading to neurotic dystrophy. This is an alternative model for the AD community to consider, although continued investigations are required to verify the model and to fill in the details.
Acknowledgement
The study was supported by NASA, The Florida Space Grant Consortium and The Community Foundation for Brevard. We thank Dr. David Tipton and Dr. Daniel Woodard of Kennedy Space Center for their support. We also thank Dr. Michael Grace for the use of his cryotome, Dr. David Carroll for frog husbandry assistance and the use of a micromanipulator, and the Florida Institute of Technology microscopy center.
References
- Wisniewski HM, Vorbrodt AW, Wegiel J, Morys J, Lossinsky AS (1990) Ultrastructure of the cells forming amyloid fibers in Alzheimer disease and scrapie. Am J Med Genet Suppl 7: 287-297.
- Yamaguchi H, Nakazato Y, Shoji M, Ihara Y, Hirai S (1990) Ultrastructure of the neuropil threads in the Alzheimer brain: Their dendritic origin and accumulation in the senile plaques. Acta neuropathologica 80: 368-374.
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: 353-356.
- Savory J, Ghribi O, Herman MM (2002) Is amyloid beta-peptide neurotoxic or neuroprotective and what is its role in the binding of metal ions? Neurobiol Aging 23: 1089-1092.
- Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26: 267-298.
- Douglas PM, Treusch S, Ren HY, Halfmann R, Duennwald ML, et al. (2008) Chaperone-dependent amyloid assembly protects cells from prion toxicity. Proc Natl Acad Sci USA 105: 7206-7211.
- Morris GP, Clark IA, Vissel B (2014) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta neuropathologica communications 2: 135.
- Benilova I, Karran E, De Strooper B (2012) The toxic abeta oligomer and Alzheimer’s disease: An emperor in need of clothes. Nat Neurosci 15: 349-357.
- Tan SY, Pepys MB (1994) Amyloidosis. Histopathology 25: 403-414.
- Hawkins PN (1988) Amyloidosis. Blood Rev 2: 270-280.
- Dickson DW (1997) The pathogenesis of senile plaques. J Neuropathol Exp Neurol 56: 321-339.
- Burnett L, Burnett B, Li B, Durrance S, Xu S (2014) A lysozyme concentration, pH, and time-dependent isothermal transformation diagram reveals fibrous amyloid and non-fibrous, amorphous aggregate species. Open J Biophys 4: 39-50.
- Bouras C, Hof PR, Giannakopoulos P, Michel JP, Morrison JH (1994) Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: A quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex. 4: 138-150.
- Chiti F, Dobson CM (2006) Protein misfolding, functional amyloid and human disease. Annu Rev Biochem 75: 333-366.
- Xu S, Bevis B, Arnsdorf MF (2001) The assembly of amyloidogenic yeast sup35 as assessed by scanning (atomic) force microscopy: An analogy to linear colloidal aggregation? Biophys J 81: 446-454.
- Xu S, Brunden KR, Trojanowski JQ, Lee VM (2010) Characterization of tau fibrillization in vitro. Alzheimers Dement. 6: 110-117.
- Xu S (2007) Aggregation drives "misfolding" in protein amyloid fiber formation. Amyloid. 14: 119-131.
- Xu S (2009) Cross-beta-sheet structure in amyloid fiber formation. J Phys Chem B 113: 12447-12455.
- De la Torre JC, Mussivand T (1993) Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol Res 15: 146-153.
- Westphalen RI, Scott HL, Dodd PR (2003) Synaptic vesicle transport and synaptic membrane transporter sites in excitatory amino acid nerve terminals in Alzheimer disease. J Neural Transm 110: 1013-1027.
- Parameshwaran K, Dhanasekaran M, Suppiramaniam V (2008) Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp Neurol. 210(1):7-13.
- Nistico R, Pignatelli M, Piccinin S, Mercuri NB, Collingridge G (2012) Targeting synaptic dysfunction in Alzheimer’s disease therapy. Mol Neurobiol 46: 572-587.
- Ohnishi S, Takano K (2004) Amyloid fibrils from the viewpoint of protein folding. Cell Mol Life Sci 61: 511-524.
- Aarli JA (2003) Role of cytokines in neurological disorders. Curr Med Chem 10: 1931-1937.
- Hardy JA, Higgins GA (1992) Alzheimer's disease: The amyloid cascade hypothesis. Science (New York, NY). 256: 184-185.
- Nordberg A (2008) Amyloid plaque imaging in vivo: Current achievement and future prospects. Eur J Nucl Med Mol Imaging 35: S46-50.
- Villemagne VL, Fodero-Tavoletti MT, Pike KE, Cappai R, Masters CL, et al. (2008) The art of loss: Abeta imaging in the evaluation of Alzheimer’s disease and other dementias. Mol Neurobiol 38: 1-15.
- Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron. 44: 181-193.
- Haataja L, Gurlo T, Huang CJ, Butler PC (2008) Islet amyloid in type 2 diabetes and the toxic oligomer hypothesis Endocr Rev 29: 303-316.
- Saric A, Chebaro YC, Knowles TP, Frenkel D (2014) Crucial role of nonspecific interactions in amyloid nucleation. Proc Natl Acad Sci USA 111: 17869-17874
- Lesne S, Kotilinek L, Ashe KH ( 2008) Plaque-bearing mice with reduced levels of oligomeric amyloid-beta assemblies have intact memory function. Neuroscience 151: 745-749.
- Caughey B, Lansbury PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26: 267-298.
- Butterfield DA, Castegna A, Lauderback CM, Drake J (2002) Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging 23: 655-664.
- Duran-Gonzalez J, Michi ED, Elorza B, Perez-Cordova MG, Pacheco-Otalora LF, et al. (2013) Amyloid beta peptides modify the expression of antioxidant repair enzymes and a potassium channel in the septohippocampal system. Neurobiol Aging 34: 2071-2076.
- Evangelisti E, Wright D, Zampagni M, Cascella R, Fiorillo C, et al. (2013) Lipid rafts mediate amyloid-induced calcium dyshomeostasis and oxidative stress in Alzheimer’s disease. Curr Alzheimer Res10: 143-153.
- Narayan P, Holmstrom KM, Kim DH, Whitcomb DJ, Wilson MR, et al. (2014) Rare individual amyloid-beta oligomers act on astrocytes to initiate neuronal damage. Biochemistry 53: 2442-2453.
- Arispe N (2004) Architecture of the alzheimer's a beta p ion channel pore. J Membr Biol 197: 33-48.
- Jang H, Connelly L, Arce FT, Ramachandran S, Kagan BL, et al. (2013) Mechanisms for the insertion of toxic, fibril-like beta-amyloid oligomers into the membrane. J Chem Theory Comput 9: 822-833.
- Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, et al. (2004) Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci 24: 10191-10200.
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, et al. (2005) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 8: 79-84.
- Klyubin I, Cullen WK, Hu NW, Rowan MJ (2012) Alzheimer's disease abeta assemblies mediating rapid disruption of synaptic plasticity and memory. Mol Brain 5: 25.
- Ferreira ST, Lourenco MV, Oliveira MM, De Felice FG (2015) Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci 9: 191.
- Kim H-E, Du F, Fang M, Wang X (2005) Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on apaf-1. Proc Natl Acad Sci USA 102: 17545-17550.
- Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, et al. (2012) Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-beta. Nature 485: 651-655.
- Um JW, Strittmatter SM (2013) Amyloid-beta induced signaling by cellular prion protein and fyn kinase in Alzheimer disease. Prion 7: 37-41
- Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457: 1128-1132.
- Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, et al. (2010) Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mglur5. Neuron 66: 739-754
- Viola KL, Klein WL (2015) Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment and diagnosis. Acta neuropathologica 129: 183-206.
- Jarosz-Griffiths HH, Noble E, Rushworth JV, Hooper NM (2016) Amyloid-beta receptors: The good, the bad and the prion protein. J Biol Chem. 291: 3174-3183.
- De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, et al. (2009) Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of abeta oligomers. Proc Natl Acad Sci USA 106: 1971-1976.
- Bitel CL, Kasinathan C, Kaswala RH, Klein WL, Frederikse PH (2012) Amyloid-beta and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model. J Alzheimers Dis 32: 291-305.
- Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, et al. (2012) An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by alzheimer's disease- associated abeta oligomers. J Clin Invest 122: 1339-1353.
- Di Scala C, Troadec JD, Lelievre C, Garmy N, Fantini J, et al. (2014) Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer beta-amyloid peptide. J Neurochem. 128: 186-195.
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, et al. (2008) Alzheimer's disease-type neuronal tau hyperphosphorylation induced by a beta oligomers. Neurobiol Aging 29: 1334-1347.
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, et al. (2003) Self-assembly of abeta(1-42) into globular neurotoxins. Biochemistry 42: 12749-12760.
- Hardy J (2009) The amyloid hypothesis for alzheimer's disease: A critical reappraisal. J Neurochem. 110: 1129-1134.
- LaFerla FM, Green KN (2012) Animal models of Alzheimer disease. Cold Spring Harb Perspect Med 2.
- Cavanaugh SE, Pippin JJ, Barnard ND. 2014. Animal models of Alzheimer disease: Historical pitfalls and a path forward. Altex 31: 279-302.
- Teich AF, Arancio O (2012) Is the amyloid hypothesis of Alzheimer’s disease therapeutically relevant? Biochem J 446: 165-177.
- Hefti F, Goure WF, Jerecic J, Iverson KS, Walicke PA, et al. (2013) The case for soluble abeta oligomers as a drug target in Alzheimer’s disease. Trends Pharmacol Sci 345: 261-266.
- Rosenblum WI (2014) Why alzheimer trials fail: Removing soluble oligomeric beta amyloid is essential, inconsistent and difficult. Neurobiol Aging 35: 969-974
- Ecroyd H, Carver JA (2008) Unraveling the mysteries of protein folding and misfolding. IUBMB Life 60: 769-774.
- Woodard D, Bell D, Tipton D, Durrance S, Burnett LC, et al. (2014) Gel formation in protein amyloid aggregation: A physical mechanism for cytotoxicity. PLoS ONE 9: e94789.
- Wisniewski HM, Vorbrodt AW, Wegiel J, Morys J, Lossinsky AS (1990) Ultrastructure of the cells forming amyloid fibers in Alzheimer disease and scrapie. Am J Med Genet Suppl 7: 287-297.
- Yamaguchi H, Nakazato Y, Shoji M, Ihara Y, Hirai S (1990) Ultrastructure of the neuropil threads in the alzheimer brain: Their dendritic origin and accumulation in the senile plaques. Acta Neuropathologica 80: 368-374.
- Nicholson C, Kamali-Zare P, Tao L (2011) Brain extracellular space as a diffusion barrier. Comput Vis Sci 14: 309-325.
- Godin AG, Varela JA, Gao Z, Danne N, Dupuis JP, et al. (2017) Single-nanotube tracking reveals the nanoscale organization of the extracellular space in the live brain. Nat Nanotechnol 12: 238-243.
- Sykova E, Nicholson C (2008) Diffusion in brain extracellular space. Physiol Rev 88: 1277-1340
- Vanharreveld A, Crowell J, Malhotra SK (1965) A study of extracellular space in central nervous tissue by freeze-substitution. J Cell Biol 25: 117-137.
- Nicholson C, Sykova E (1998) Extracellular space structure revealed by diffusion analysis. Trends Neurosci 21: 207-215.
- Thorne RG, Nicholson C. 2006. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci USA 103: 5567-5572.
- Zhang H, Verkman AS (2010) Microfiberoptic measurement of extracellular space volume in brain and tumor slices based on fluorescent dye partitioning. Biophys J 99: 1284-1291.
- Rusakov DA, Kullmann DM (1998) Geometric and viscous components of the tortuosity of the extracellular space in the brain. Proc Natl Acad Sci USA 95: 8975-8980
- Patlak CS, Fenstermacher JD (1975) Measurements of dog blood-brain transfer constants by ventriculocisternal perfusion. Am. J Physiol 229: 877-884.
- Archie GE (1942) The electrical resistivity log as an aid in determining some reservoir characteristics. Trans Am Inst Mining, Metallurgical Petroleum Engineers Inc., pp: 54-62.
- Nicholson C, Rice ME (1986) The migration of substances in the neuronal microenvironment. Ann N Y Acad Sci 481: 55-71.
- Kume-Kick J, Mazel T, Vorisek I, Hrabetova S, Tao L, et al. (2002) Independence of extracellular tortuosity and volume fraction during osmotic challenge in rat neocortex. J Physiol 542: 515-527.
- Pfeuffer J, Dreher W, Sykova E, Leibfritz D (1998) Water signal attenuation in diffusion-weighted h-1 NMR experiments during cerebral ischemia: Influence of intracellular restrictions, extracellular tortuosity and exchange. Magn Reson Imaging 16: 1023-1032.
- Mota M, Teixeira JA, Keating JB, Yelshin A (2004) Changes in diffusion through the brain extracellular space. Biotechnol Appl Biochem 39: 223-232.
- Abbott NJ (2004) Evidence for bulk flow of brain interstitial fluid: Significance for physiology and pathology. Neurochem Int 45: 545-552.
- Brinker T, Stopa E, Morrison J, Klinge P (2014) A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 11: 10.
- Rapoport SI (1978) A mathematical model for vasogenic brain edema. J Theor Biol 74: 439-467.
- Damkier HH, Brown PD, Praetorius J (2013) Cerebrospinal fluid secretion by the choroid plexus. Physiol Rev 93: 1847-1892.
- Ichimura T, Fraser PA, Cserr HF (1991) Distribution of extracellular tracers in perivascular spaces of the rat brain. Brain Res 545: 103-113.
- Rosenberg GA, Kyner WT, Estrada E (1980) Bulk flow of brain interstitial fluid under normal and hyperosmolar conditions. Am. J. PhysioL238: F42-49.
- Bradbury MW, Cserr HF, Westrop RJ (1981) Drainage of cerebral interstitial fluid into deep cervical lymph of the rabbit. Am J Physiol 240: F329-336.
- Cserr HF, Cooper DN, Suri PK, Patlak CS (1981) Efflux of radiolabeled polyethylene glycols and albumin from rat brain. Am J PhysioL240: F319-328.
- Szentistvanyi I, Patlak CS, Ellis RA, Cserr HF (1984) Drainage of interstitial fluid from different regions of rat brain. Am J Physiol 246: F835-844.
- Ohata K, Marmarou A (1992) Clearance of brain edema and macromolecules through the cortical extracellular space. J Neurosurg 77: 387-396.
- Pullen RG, DePasquale M, Cserr HF (1987) Bulk flow of cerebrospinal fluid into brain in response to acute hyperosmolality. Am J Physiol 253: F538-545.
- Mueggler T, Meyer-Luehmann M, Rausch M, Staufenbiel M, Jucker M, et al. (2004) Restricted diffusion in the brain of transgenic mice with cerebral amyloidosis. Eur J Neurosci 20: 811-817.
- Mao-Draayer Y, Braff SP, Nagle KJ, Pendlebury W, Penar PL, et al. (2002) Emerging patterns of diffusion-weighted mr imaging in Creutzfeldt-Jakob disease: Case report and review of the literature. AJNR Am J Neuroradiol 23: 550-556.
- Sykova E Vorisek I, Antonova T, Mazel T, Meyer-Luehmann M J (2005) Changes in extracellular space size and geometry in app23 transgenic mice: A model of Alzheimer’s disease. Proc Natl Acad Sci USA 102: 479-484
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, et al. (2000) Clearance of Alzheimer’s amyloid-ss (1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106: 1489-1499
- Lam FC, Liu R, Lu P, Shapiro AB, Renoir JM, et al. (2001) Beta-amyloid efflux mediated by p-glycoprotein. J Neurochem 76: 1121-1128.
- Weller RO (1998) Pathology of cerebrospinal fluid and interstitial fluid of the CNS: Significance for Alzheimer disease, prion disorders and multiple sclerosis. J Neuropathol Exp Neurol 57: 885-894.
- Aguzzi A, O'Connor T (2010) Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat Rev Drug Discov 9: 237-248.
- Suzuki Y, Nakamura Y, Yamada K, Igarashi H, Kasuga K, et al. (2015) Reduced CSF water influx in Alzheimer's disease supporting the beta-amyloid clearance hypothesis. PLoS ONE 10: e0123708
- Janmey PA (1982) Kinetics of formation of fibrin oligomers. I. Theory. Biopolymers 21: 2253-2264.
- Weisel JW (2007) Structure of fibrin: Impact on clot stability. J Thromb Haemost 5: 116-124.
- Greenwald J, Riek R (2010) Biology of amyloid: Structure, function and regulation. Structure 18: 1244-1260.
- Bramanti E, Benedetti E, Sagripanti A, Papineschi F, Benedetti E (1997) Determination of secondary structure of normal fibrin from human peripheral blood. Biopolymers 41: 545-553.
- Kranenburg O, Bouma B, Kroon-Batenburg LM, Reijerkerk A, Wu YP, et al. (2002) Tissue-type plasminogen activator is a multi-ligand cross-beta structure receptor. Curr Biol 12: 1833-1839.
- Grabowski ZR, Rotkiewicz K, Rettig W (2003) Structural changes accompanying intramolecular electron transfer: Focus on twisted intramolecular charge-transfer states and structures. Chem Rev 103: 3899-4032.
- Akers WJ, Cupps JM, Haidekker MA (2005) Interaction of fluorescent molecular rotors with blood plasma proteins. Biorheology 42: 335-344.
- Haidekker MA, Theodorakis EA (2010) Environment-sensitive behavior of fluorescent molecular rotors. J Biol Eng 4: 11.
- Breit S (2013) Amyloid fibers - Mechanisms of formation and potential role in neuronal death. (Melboure, FL): Florida Institute of Technology.
- Rogers J, Morrison JH (1985) Quantitative morphology and regional and laminar distributions of senile plaques in Alzheimer’s disease. J Neurosci 5: 2801-2808
- Balthazar ML, Yasuda CL, Pereira FR, Pedro T, Damasceno BP, et al. (2009) Differences in grey and white matter atrophy in amnestic mild cognitive impairment and mild Alzheimer’s disease. Eur J Neurol 16: 468-474.
- Brun A, Englund E (1986) A white matter disorder in dementia of the Alzheimer type: A pathoanatomical study Ann Neurol 19: 253-262.
- Roher AE, Weiss N, Kokjohn TA, Kuo YM, Kalback W, et al (2002) Increased a beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer's disease. Biochemistry 41: 11080-11090
- Sachdev PS, Zhuang L, Braidy N, Wen W (2013) Is Alzheimer's a disease of the white matter? Curr Opin Psychiatry 26: 244-251
- Amlien IK, Fjell AM (2014) Diffusion tensor imaging of white matter degeneration in Alzheimer's disease and mild cognitive impairment. Neuroscience 276: 206-215.
- Serra L, Cercignani M, Basile B, Spano B, Perri R, et al. (2012) White matter damage along the uncinate fasciculus contributes to cognitive decline in AD and DLB. Curr Alzheimer Res 9: 326-333
- Collins-Praino LE, Francis YI, Griffith EY, Wiegman AF, Urbach J, et al. (2014) Soluble amyloid beta levels are elevated in the white matter of Alzheimer's patients, independent of cortical plaque severity. Acta Neuropathologica communications 2: 83.
- Zhu YJ, Zeng T, Zhu YB, Yu SF, Wang QS, (2008) Effects of acrylamide on the nervous tissue antioxidant system and sciatic nerve electrophysiology in the rat. Neurochem Res 33: 2310-2317.
- Benzon HT, Gissen AJ, Strichartz GR, Avram MJ, Covino BG (1987) The effect of polyethylene glycol on mammalian nerve impulses. Anesth Analg 66: 553-559.
- Morse RP, Evans EF (2003) The sciatic nerve of the toad Xenopus laevis as a physiological model of the human cochlear nerve. Hear Res 182: 97-118.
- Peters R, Peters J, Tews KH, Bahr W (1974) A microfluorimetric study of translational diffusion in erythrocyte membranes. Biochim Biophys Acta 367: 282-294.
- Braeckmans K, Peeters L, Sanders NN, De Smedt SC, Demeester J (2003) Three-dimensional fluorescence recovery after photobleaching with the confocal scanning laser microscope. Biophys J 85: 2240-2252.
- Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW (1976) Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J 16: 1055-1069.
- Soumpasis DM (1983) Theoretical analysis of fluorescence photobleaching recovery experiments. Biophys J. 41: 95-97
- Seiffert S, Oppermann W (2005) Systematic evaluation of frap experiments performed in a confocal laser scanning microscope. J Microsc 220: 20-30
- Miyawaki A (2011) Proteins on the move: Insights gained from fluorescent protein technologies. Nat Rev Mol Cell Biol 12: 656-668.
- Trembecka DO, Kuzak M, Dobrucki JW (2010) Conditions for using frap as a quantitative technique--influence of the bleaching protocol. Cytometry A 77: 366-370.
- Hladky SB, Barrand MA (2014) Mechanisms of fluid movement into, through and out of the brain: Evaluation of the evidence. Fluids Barriers CNS 11: 26.
- Buishas J, Gould IG, Linninger AA (2014) A computational model of cerebrospinal fluid production and reabsorption driven by starling forces. Croat Med J 55: 481-497.
- Stine WB, Dahlgren KN, Krafft GA, LaDu MJ (2003) In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem 278: 11612-11622
Relevant Topics
- Advanced Parkinson Treatment
- Advances in Alzheimers Therapy
- Alzheimers Medicine
- Alzheimers Products & Market Analysis
- Alzheimers Symptoms
- Degenerative Disorders
- Diagnostic Alzheimer
- Parkinson
- Parkinsonism Diagnosis
- Parkinsonism Gene Therapy
- Parkinsonism Stages and Treatment
- Stem cell Treatment Parkinson
Recommended Journals
Article Tools
Article Usage
- Total views: 3783
- [From(publication date):
August-2017 - Aug 20, 2025] - Breakdown by view type
- HTML page views : 2891
- PDF downloads : 892