Assessing Stability, Durability, and Protein Adsorption Behavior of Hydrophilic Silane Coatings in Glass Microchannels
Received: 06-Apr-2016 / Accepted Date: 18-Apr-2016 / Published Date: 25-Apr-2016 DOI: 10.4172/2155-9872.1000318
Keywords: Non-specific adsorption; Microfluidic separations; Protein-surface interactions; Surface coatings; Silane coatings
304324Introduction
Advances in microfabrication technology, and the consequent ability to precisely control geometrical features down to the submicron scale, have enabled the development of novel micro- and nanofluidic platforms. This new generation of miniaturized devices allows for faster analysis on smaller sample volumes, integration of multiple functionalities on the same platform, and better sensitivity at a lower cost. Furthermore, such devices have also enabled novel applications and methods that exploit the physics of fluids in micro and nanoconfinements, from novel separation modalities [1,2], to fluid pumping by diffusio-osmosis [3], to energy conversion [4].
As more research focuses on medical and diagnostic applications involving micro- and nanofluidic devices, the issue of non-specific adsorption of biomolecules at the channel surface is gaining increasing attention [5-7]. In the case of micro-scale electrokinetic separations, for example, unwanted wall-sample interactions lead to non-uniform charge density on the inner capillary wall and, thus, to non-uniform electroosmotic flow profiles [6,8], subsequently causing sample dispersion and degradation of separation resolution Furthermore, such interactions affect peak shape and electromigration times, potentially tainting analysis results [7,9,10]. Finally, loss of sample and degradation of device functionality are additional pressing concerns.
In general, the main mechanisms of nonspecific interactions between biomolecules and solid surfaces are electrostatic (ionic), hydrophobic and hydrogen bond interactions [10,11]. Electrostatic interactions arise from Coulombic attraction or repulsion between charged groups on a biomolecule, and are highly affected by pH. Hydrophobic interactions can cause biomolecules to aggregate, and also play a major role in biomolecular adsorption from water onto hydrophobic surfaces [11,12]. Among biomolecules, protein adsorption to surfaces poses a unique challenge, apparent through the plethora of attention in the literature [13-18]. Proteins often denature upon adsorbing and become very difficult to remove from the walls, limiting both device reliability and reusability [11,19,20].
A straightforward way to control protein surface adsorption is to use buffers at pH extremes, to either operate above the isoelectric point of proteins, or suppress ionization of silanol groups at the channel walls [10,21]. Under these conditions, electrostatic repulsion forces prevent proteins from adsorbing to the wall. However, the abundance of OHions at pH>11, and H+ at pH<2 can result in the generation of large currents and Joule heating. In addition, the buffering power of most electrolytes is very poor at these pH extremes [21]. More importantly, these conditions are not suitable to study the activity of proteins in native environments. More sophisticated methods to eliminate or reduce adsorption include studies to control the wall potential by means of an external electric field [22,23], or using additives in the buffer solution to compete for cation-exchange sites on the wall [24-27]. However, by far the most popular method to mitigate adsorption is the use of surface coatings [27-30]. Not only is their use widespread in microscale devices [27,31-33]: research to develop new types of chemical coatings is also flourishing [34,35].
Ideally, surface coatings should possess a variety of properties including: biocompatibility, hydrophilicity, electrical neutrality, hydrogen bond acceptance, self-repulsion, and conformational flexibility [36,37]. In recent years, a number of researchers have synthesized and characterized many surface coatings [9,35,38-40] intended to prevent analyte adsorption within micro- and nanofluidic devices [27,31,32]. However, there is no comparative quantitative data between promising coatings, and meaningful comparisons of data between different experimental studies are not always possible [5,39]. This paper aims to fill this gap by providing a quantitative comparative analysis of surface coatings for the purpose of assessing their properties and behaviors in controlled conditions relevant to microfluidics, specifically for microfluidic based protein separations. We chose to use bovine serum albumin (BSA) and immunoglobulin G (IgG) as model proteins, given their propensity to adsorb to a variety of different surfaces, and the fact that they are the two most abundant proteins in blood. In addition, the behavior of BSA and IgG may be used as an indication for respectively, other serum or globular proteins (BSA) and antibodies (IgG).
We chose to assess the performance of some of the most common surface coatings: two hydrophilic (2-[methoxy(polyethyleneoxy)] propyl trimethoxysilane (MPEG) and 3-mercaptopropyl (3-MPS) silane), two ionic (a zwitterionic sultone derived silane (ZS), and a zwitterionic phosphate derived silane (ZP)), and 3-aminopropyl (3- APS) silane. We compare these coatings with regards to: a) the ability to successfully silanize planar microfluidic channels (since the procedures we propose are adapted from existing recipes used to coat various silica substrates such as glass slides and beads), b) stability and durability over time via zeta potential (ζ) measurements, and c) antifouling performance through protein interactions.
Materials and Methods
Materials
We used two types of fused silica, isotropically-etched microfluidic devices for this study: simple cross (N, W, S: 5 mm long, E: 30 mm long), 20 μm deep × 50 μm wide channels (Dolomite Ltd, UK) for competitive electrophoresis injection experiments, and straight custom-fabricated 20 μm deep × 25 mm long channels for continuous flow experiments (Figure 1a and b respectively). We also used plain Borosilicate coverslips (Corning, Catalog Number 2865-22) as a control surface with no continuous flow or confinement.

Figure 1: Schematic depiction of microchannel geometries used in this study. Cross channels are used for electrophoresis injection experiments, while straight channels are used for continuous flow measurements. All channels were fabricated from fused silica (Dolomite Ltd., UK).
Chemicals and reagents for surface coating
Synthesis: All chemicals and reagents were purchased from Sigma- Aldrich with the exception of MPEG-silane which was purchased from Gelest. 2-(dimethyl(3-(trimethoxysilyl)propyl)ammonio)ethane-1- sulfonate (zwitterionic sultone silane, ZS-silane) was prepared by the reaction of 1,3-propanesultone with N,N-dimethyl-3-(trimethoxysilyl) propan-1-amine in acetone according to the procedure outlined in Ref. [41]. 2-(dimethyl(3-(trimethoxysilyl)propyl)ammonio)ethyl ethyl phosphate (zwitterionic phosphate silane, ZP-silane) was prepared in two steps according to the procedure outlined in Ref. [42].
Silanization on glass slides: Borosilicate coverslips (Corning, Catalog Number 2865-22) were modified by immersion in a solution containing 3-4% w/v silane (in ethanol, water or toluene) and incubated at 40°C for 18 hours. The coverslips were then removed from the incubation solution, washed with ethanol and water, dried, and stored at ambient temperature [43]. MPEG-silane and zwitterionic silanes were incorporated using methods reported in Ref. [42,44].
Silanization in channels: MPEG-silane and ZS-silane were prepared as 20 mL of a 4% w/v solution in deionized water. 10 μL of the desired silane solution was added to a single well of the microchannel. Upon full channel wetting (as confirmed by light microscopy), the remaining wells were filled with 10 μL of the silane solution and the entire microchannel chip was immersed in the silane solution and incubated at 80°C for 18 hours. The microchannel chip was then removed from the silane solution and the exterior washed extensively with DI water. The microchannel was rinsed by inducing an electrokinetic flow (applied voltage V=100 V) using DI water for thirty minutes. 3-APS, 3-MPS and ZP-silane were prepared as 20 mL of a 2% to 4% w/v solution of the silane in EtOH (3-APS and ZP-silane) or toluene (3-MPS) and 10 μL of the silane solution was added to a single well of the microchannel. Upon full channel wetting (as confirmed by light microscopy), the remaining wells were filled with 10 μL of the silane solution and the entire microchannel chip was immersed in the silane solution and incubated at room temperature for 3-APS and ZPsilane or 45°C for 3-MPS, overnight. After modification was complete, the microchannel chip was removed from the silane solution and the exterior of the chip was washed with EtOH. The microchannel was then flushed by inducing an electrokinetic flow of EtOH, 1:1 EtOH:DI water, and DI water in successive 30 minute periods (applied voltage V=100 V). All microchannels were stored in DI water at ambient temperature until use.
Protein solutions and slide incubation: We incubated modified coverslips in 10 mM sodium borate buffer (pH 9) with either: a) FITClabelled bovine serum albumin (BSA), or b) immunoglobulin G (IgG). Figure 2 depicts electrostatic potential maps for both proteins. Protein concentrations for BSA and IgG were prepared in concentrations of 1 mg/mL and 2 mg/mL, respectively. We used all solutions immediately after preparation to prevent protein degradation. For adsorption experiments, we immersed glass slides in the appropriate protein solution and allowed it to incubate for 24 hours in the dark, to prevent bleaching. Before performing fluorescence imaging measurements, all glass slides were soaked twice in DI water for 2 hours with agitation, to remove excess protein.

Figure 2: Electrostatic potential map for a) BSA and b) IgG. Blue regions represent negative charges, while red represent positive.
Data collection methods
Fluorescence imaging: We measured all fluorescence data by recording images with an inverted epifluorescence microscope (Olympus IX70, Olympus, Inc.) fitted with a 20X water immersion objective lens (0.45 NA, Olympus, Inc.). Illumination from a 200 W Hg-arc lamp was filtered with a FITC fluorescence filter cube (Omega, Inc.) containing excitation and emission filters and a dichroic mirror matched to the fluorescence spectrum of FITC. We recorded images using a back illuminated EMCCD camera (Ixon Ultra897, Andor Technology) with a 512 × 512 pixel array and 16-bit digitization to a PC. Frame rate (10 to 20 Hz) and exposure time (0.05 to 0.1 s) varied, depending on the channel depth and analyte, to maximize signal-to-noise ratio. We performed background subtraction and flatfield correction on all images using custom Matlab (The MathWorks) programs, to further enhance signal to noise ratio [45].
To measure protein adsorption on slides, after silanization and protein incubation as described above, we collected one image at three separate locations. For each location, we computed the average fluorescence value by randomly selecting ten pixels. For each combination of surface coating and protein, we performed experiments on two slides. Results, in terms of average absolute fluorescence and pooled variance, are shown in Figure 3.

Figure 3: Adsorption experiments on modified coverslips, incubated in 10 mM sodium borate buffer (pH 9) with: a) 1 mg/mL FITC-labelled BSA, and b) 2 mg/ mL IgG. In both cases, PEG and zwitterionic coatings incur the least amount of protein adsorption.
For fluorescence data in straight channels, after surface coatings were applied to the channel as described above, the channels were rinsed with DI water, and then filled with 10 mM sodium borate buffer solution (pH=9). We applied an external voltage, V=300 V (Keithley 2410), through platinum electrodes (Omega Eng. Inc., Stamford, CT) to establish an electroosmotic flow. Next, protein solution was added to the inlet ports and fluorescence values were monitored over time, while the protein solution was electrokinetically driven in the channels. After fluorescence reached its maximum value (different for each coating and protein combination), channels were rinsed with a continuous flow of 10 mM sodium borate buffer. We also monitored fluorescence during this phase. Fluorescence values displayed in Figure 4 show the average fluorescence within the channel after background and flat field correction.

Figure 4: Normalized residual fluorescence intensity measurements after sodium borate buffer flushing of a) BSA and b) IgG through a straight microfluidic channel. The light and dark bars represent, respectively, the fluorescence values at the beginning and after 30 minutes of flushing. Fluorescence values are corrected by subtracting background and dividing by flat field values.
Current monitoring: Prior to all measurements, we electrokinetically drove deionized water into the straight channels (Figure 1b) by applying a voltage V=100 V (Keithley 2410) until the current, monitored using a second electrometer (Keithley 2410) stabilized. Next, the channels were filled with a 9 mM sodium borate buffer solution (SBX). After current stabilization, we replaced the 9 mM SBX solution in reservoir 1 by 10 mM SBX, and recorded the increase in current, as the channel filled with the higher concentration buffer. We repeated this process 3-4 times for each coating. To derive the zetapotential, we solve for bulk average electroosmotic velocity, vEOF, from current monitoring using a custom Matlab script in which the length of the channel is divided by the time to fill the channel. From this value, the zeta-potential is found using the classic Helmholtz-Smoluchowski equation:
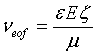
where μ is viscosity (8.90 × 10-4 Pa for water), E is the applied electric field, and ζ is the zeta-potential [46]. For the dielectric constant ε, we use the value of water, given the very low molarity of the buffers used.
Microfluidic injections: Microfluidic injections are typically used to assess the electroosmotic mobility of fluorescently tagged species. This technique can also be used to study adsorption of fluorescent species by, for example, examining the fluorescent residue that remains on the channel wall once a fluorescently-tagged protein travels down the channel, or in our case, measuring the peak intensity over time after successive injections. Figure 1a shows a schematic of the channel geometry and our naming convention for each well: N (northern), S (southern), W (western) and E (eastern). Electrical potentials were applied at these wells using platinum electrodes (Omega Eng. Inc., Stamford, CT), connected to a high voltage power supply (LabSmith HVS448). The pre-programmed voltage scheme for sample loading and injection was designed following the recommendations of [47]. Briefly, during the loading phase, the sample solution is placed in the N well and electrodes in the N, W, and E wells are set to positive voltages while the S well is grounded, resulting in electrokinetic flow from all wells towards S. For the injection step, the applied voltages are then switched (W, N, S at high, E at ground), and the sample is injected along the E channel. The electric field during injections is 78.8 V/m. Injection data is recorded 10 mm downstream from the injection point with a high sensitivity EMCCD camera (Andor iXon), fitted to a 20X objective. The resulting electropherograms display fluorescence intensity over time, as plugs corresponding to different analytes pass through the detection point (For example: Figure 5).

Figure 5: Electropherogram of FITC-labeled injections in a ZS-coated microfluidic chip (20 μm deep glass cross channel). Trials chosen to be representative of the trend. Protein sample is suspended in 10 mM sodium borate buffer (pH 9) and composed of: a) IgG, b) BSA, c) IgG and BSA.
For each experiment (Figures 5 and 6), the same sample is injected 20-30 times, by applying the voltage sequence inject - load - inject, with the load step long enough to allow for the previously injected plug to flow towards the S well. In these electropherograms, a net reduction in peak intensity and area indicates protein adsorption.

Figure 6: Electropherogram of IgG-FITC in a PEG coated (20 μm glass cross channel). Intensity (AU) over Elution time as shown. Trials chosen to be representative of the trend. Area under the curve was calculated for each trial and then percent area is displayed in reference to the initial trial (area max).
Results
Adsorption measurements on glass slides
We first measured the degree of protein absorption of the five chosen surface coatings (ZS, PEG, ZP, 3-MPS and 3-APS) on glass slides, using FITC-labeled BSA and IgG as model proteins. These surface coatings were chosen from an array of 7 previously analyzed [43]. Specifically, we chose MPEG because it is the proven standard hydrophilic silane coating, and is a universally well-known to prevent surface adsorption. ZS and ZP were chosen as possible zwitterionic candidates (hydrophilic with neutral opposing ionic charges/forces), given that zwitterions are known to help prevent protein adsorption. 3-MPS and 3-APS were both chosen as positive controls, but through different mechanisms of plausible adsorption [48,49]. 3-MPS was selected as a positive control for protein binding via disulfide linkages over 3-APS, which only provides charge based interactions.
Based on the data shown in Figure 3, which shows the residual fluorescence on the glass slide after protein adsorbed for 24 hours and was subsequently washed away with DI water. As expected, simple hydrophilic (MPEG) and zwitterionic coatings repelled proteins best, whereas hydrophilic coatings (3-MPS, 3-APS) were not effective at preventing adsorption, potentially due to the aforementioned disulfide linkages with 3-MPS or carboxylate-salt interactions between the protein c-terminus and 3-APS. The MPEG coating exhibited very low adsorption for both BSA and IgG, most likely because the formation of a denser hydration layer at the surface [50]. Zwitterions have the advantage of containing both positive and negative functional groups [51], which are responsible for ionic repulsive forces. In agreement with previous work [41], the heat-treated ZS coating resulted in the least residual fluorescence for BSA, as well as a very low value for IgG. Heat treatment is hypothesized to generate a more stable coating matrix and side chain connections between silanol groups, thus resulting in higher stability. ZP performed better than the control sample for both BSA and IgG, also in agreement with previous work [42].
Stability of surface coatings
To assess the stability over time of surface coatings we performed current monitoring experiments over a period of 6 weeks (Figure 3). In microfluidics, current monitoring is a procedure that is typically used to measure the channel zeta-potential. The zeta-potential, ζ is a parameter that characterizes the surface potential at the shear plane of the channel, and thus can be used to characterize electroosmotic flow. It is further useful in studying adsorption, because analyte adsorption at the walls will change the initial surface, and consequently the value of the zeta-potential. Thus, ζ can be used as an effective indicator of surface properties. Each ζ-potential value presented in Figure 7 represents the average of at least 3 measurements performed at each time point. We also calculated the average zeta potential over the 6 weeks measurements, ζavg, shown in the table to the right of Figure 7. Assuming the same statistical distribution of the error for each measurement, we compute the variance by the pooled variance method [52]. Finally, we estimate the variability in zeta potential defining Δζavg=(ζmax − ζmin)/ζmax. We note that potential complications for all coatings is that the electrokinetic flow may be causing shearing of the surface coatings, but current monitoring measurements are only performed for a small percentage of the time as compared to the 6 weeks that they were subjected to standard temperature and pressure (STP) conditions.

Figure 7: Absolute value of zeta potential measurements derived from current monitoring experiments during a period of six weeks. Each data point is the average of 3-4 measurements, with the error bars representing the standard deviation.
As expected, the (unmodified) control chip has the highest negative zeta potential [53]. The exposed silanol groups ionize when in contact with an aqueous solution at high pH, resulting in a high negative net surface charge. The control chip was the most stable over time, however with a surprisingly larger than expected variance Δζavg=9%. We attribute this non-zero variance over time to dissolution of the surface from – OH groups [54] or from chemicals from the ambient atmosphere that attach and change the makeup of the surface [55].
The MPEG coating exhibited the lowest value of ζavg over the duration of the six-week experiment. Because the zeta potential was not consistently increasing over time, we attribute the large variation of zeta potential (Δζavg=54%) to the low absolute ζ-potential value and thus relatively large contribution of experimental errors that are inherent with such a low measurement value. Changes in ζ-potential could be due to oxidative effects on MPEG [37,44,56] induced by electrolysis. However, no electrolytic bubbles were observed, and the external field was a constant 100 V, so we assume that the MPEG coating did not degrade, and instead the variability is due to experimental error.
3-MPS is shown to be very stable, rivaling the stability of the uncoated channel with a Δζavg=12% (Figure 3). One possible explanation of this stability is disulfide formation between MPS moieties in the coating. If this is occurring, then silane hydrolysis would not result in full delamination, and thus the channel would retain its properties over an extended period of time.
Our results for channels coated with ZS do not correlate well with literature findings. Our data shows unstable behavior, with large variation in zeta-potential values over the 6 weeks, Δζavg=54%, and a large spread in each single measurement, with each measurement decreasing in value. We hypothesize that in our case, full polymerization and formation of a stable monolayer was not achieved, and instead single point silanization may have occurred, with concomitant oligomerization resulting in island morphologies and multi-layer formation. Note that because the zeta potential absolute value was between 40-70 mV, and it was monotonically decreasing over time, we do not believe that user and experimental-error contributed to the large variance.
ZP was the most unstable, with the ζ-potential at the end of 6 weeks approaching that of an uncoated channel, suggesting that the coating completely degraded. Authors in Ref. [51] noted that ZPlike structures form densely packed hydration layers, binding water molecules more strongly than PEG. However, past work has dealt with the case of “static” surfaces that is in the absence of flow. Work by Ref. [42], using the exact same phosphate coating as the one in this study, found similar positive changes in contact angle over time in phosphate buffer, indicating degradation. The changes were attributed to weakly bound silanes diffusing out from the system. In light of these findings, we hypothesize that our observed behavior of ZP coating is the result of interactions in the presence of sodium borate buffer that, in the course of our measurements, remove weakly bound silanes via ionic interactions or hydrolyze the phosphodiester bond of the functional chain, thus shifting charge neutrality and density.
Adsorption measurements in continuous flow
We next assessed the adsorption of proteins within channels with flow, since fluid flow may change the effects of adsorption as compared to static glass slide measurements. To do so, we monitored adsorption during protein flow and subsequent aqueous-based flushing of both BSA and IgG solutions on various coated microchannels (Figure 4).
As mentioned in the introduction, proteins may be reversibly attracted to hydrophilic surfaces through electrostatic forces. These interactions may lead to irreversible conformational changes and stretching of the proteins across the surface, which results in more electrostatic interactions [57]. Factors affecting such adsorption include: 1) surface coating density, which may generate an entropic and steric environment which is favorable/unfavorable for further attachment; 2) time dependence of the process of permanent adsorption, which is directly dependent on both flow velocity and the total time proteins are flushed through the channel [58]; 3) protein concentration (we used 1 mg/ml, based on results reported in Ref. [36,59]); and 4) washing procedures, which may remove protein not permanently bound, again dependent on both flow velocity and the total time of flushing.
The time t=0 in Figure 7 corresponds to the point where we started flushing the channel with buffer solution, after exposing the channels to a continuous solution of proteins for 20-30 min. During rinsing, fluorescence was measured at regular intervals, and the process was stopped when its value came as close as possible to the initial intensity. For each coating, this time was chosen to allow for the fluorescence signal to reach its maximum value and stabilize (Table 1). Since the coating affects zeta potential and thus electroosmotic flow mobility, for different coatings the tagged proteins take a different amount of time to enter and fill the channel. It is interesting to note, how the different coatings perform at t=0, with e.g., MPEG showing the largest fluorescence value at the beginning of flushing in case of BSA, but going down to almost zero values by the end of flushing, something that could not be assessed with simple glass coverslip experiments.
| Peak FL BSA |
Residual FL BSA |
Peak FL IgG |
Residual FL IgG |
Type | Structure | |
|---|---|---|---|---|---|---|
| Control | 0.727 (7.5 min) |
0.0006 (0%) |
1.14 (20 min) |
0.329 (29%) |
Reference | |
| MPEG | 0.96 (30 min) |
0.02 (2.1%) |
1.04 (15 min) |
0.13 (12.5%) |
Hydrophilic Experimental |
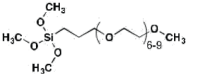 |
| ZS | 0.7 (20 min) |
0.02 (2.8%) |
1.01 (30 min) |
0.05 (4.9%) |
Zwitterionic Experimental |
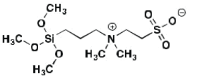 |
| 3-APS | 0.77 (15 min) |
0.036 (4.6%) |
1.1 (20 min) |
0.397 (36.1%) |
Cationic Control |
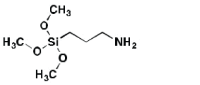 |
| ZP | 0.96 (20 min) |
0.05 (5.2%) |
1.03 (15 min) |
0.132 (12.8%) |
Zwitterionic Experimental |
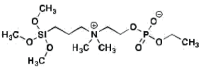 |
| 3-MPS | 0.87 (7.5 min) |
0.084 (9.65%) |
1.14 (20 min) |
0.72 (63.2%) |
Fouling Control |
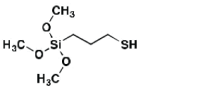 |
Table 1: Displays relevant numerical data from antifouling experiments with IgG-FITC and BSA-FITC. Peak FL is the peak fluorescence for the trial, residual FL is remainder after 30 min. “Type” corresponds to role in the study.
In terms of experiments with IgG (Figure 4b and Table 1), we find stronger fouling characteristics than BSA for all coatings considered, as expected from glass slide experiments (Figure 3). Like for the coverslips, MPEG, ZP and ZS have the lowest values of residual fluorescence during rinsing. However, contrary to the glass slide experiments, it is 3-MPS, and not the bare channel, that has the largest value of residual fluorescence, similar to the BSA case.
We posit that the difference we observe in glass coverslips with respect to microfluidic channels stems from time-dependent and flow-dependent behavior of the adsorption process, which becomes apparent only in the latter experiments. In uncoated channels, entropic interactions with the ionized silanols on glass are expected to be a contributing mechanism for protein adsorption [44], with electrostatic interactions associated with the positive BSA sites and the hydrophilic glass surface [60]. The relative ease with which protein was removed from bare glass microchannels may have resulted from the large EOF velocity, due to the large zeta-potential (Figure 7). The EOF velocity may also have hindered attachment of BSA, compared to what is expected with glass slides [61]. In the case of IgG, the uncoated channel showed a 33% residual fluorescence with respect to peak value after 30 minutes. According to Ref. [60], IgG adsorption on borosilicate vials is driven mainly by electrostatic forces, which in turn are the product of pH and ionic forces. Thus, the combination of sodium borate buffer (pH 9) and exposed glass silanols is expected to promote IgG fouling in uncoated channels, in spite of the large EOF (Figure 7). The difference between the adsorption profiles of BSA and IgG from coverslips to channels illustrates the importance of experimental conditions (Figure 3). Specifically, flow velocity can affect BSA adsorption, while the choice of buffer clearly affects IgG adsorption.
Because of the low zeta-potential, MPEG results in a slower EOF velocity for the same applied voltage. As a consequence, the behavior we observe is diffusion limited and matches static coverslip results well. ZS and ZP coatings show excellent anti-fouling properties in all cases. These results are not surprising considering the strong hydration layer formed and described also in Ref. [51], along with potentially additional electrostatic and steric mechanisms described by Ref. [44], and results from other studies [62,42]. However, changes in zeta potential over time (Figure 7) suggest that the quality of the coating may deteriorate with time. Both 3-MPS and 3-APS as expected had the worst performance, potentially due to disulfide linkages with 3-MPS or carboxylate-salt interactions between the protein c-terminus and 3-APS.
Adsorption measurements in electrokinetic flow
In consideration of data from all these experiments, we chose to further analyze the behavior of ZS and MPEG during electrokinetic separation injections of proteins in cross channel glass chips. The purpose behind evaluating competitive injections was to assess coating performance in discrete trials and with multiple proteins, as opposed to continuous exposure to a single protein. Furthermore, such injections are a cornerstone of microfluidic capillary electrophoresis, which has, through the years, been highlighted as a possible cheaper, faster, alternative method for performing separations of a variety of complex mixtures [63-70].
Figure 5 shows a series of injections through a ZS coated cross channel using sodium borate buffer solution containing FITC-labeled IgG and BSA. The first two panels, Figure 5a and b, show that when BSA or IgG are injected alone, changes between serial injections are negligible. The arrival of the BSA peak in Figure 5b has a very small delay, which could indicate that over one injection the BSA absorbed to the channel, slightly changing the zeta potential, and thus arriving at a slightly later time. However, the results seem to be well within experimental error.
Figure 5c shows a series of electropherograms with both IgG and BSA injected at the same time. Here, the BSA peak changes drastically between the first two injections, with a major decrease in overall intensity, as well as a major shift in arrival time. The IgG peak intensity is also reduced over time, more significantly than shown in Figure 5a. The marked decrease in fluorescence as the measurements progress in time is an indication of sample adsorption at the channel walls: a process that seems to continue throughout the experiment. The absence of tailing suggests a practically irreversible process [10], which, contrary to experiments in glass slides (Figure 3), mostly affects BSA. It is known that, in the presence of multiple analytes, there is a competitive behavior for available binding sites, leading to one analyte prevailing, based on the mechanism of adsorption for the specific environment [59]. Further, competitive protein experiments are known to show protein dominance based on diffusivity [58] and it has been established that on hydrophilic media such as glass, albumin tends to adsorb first, which is then followed by IgG, based on size differences [71]. Our results also clearly indicate these phenomena at play, which result in drastic differences between single vs. multiple analyte electropherograms, and ultimately separation efficiency. Estephan et al. [44] outlined a mechanism for adsorption via ionic forces where zwitterionic sultone (and like) do not allow the removal of counterion base pairs from the protein and surface respectively. This therefore inhibits attachment at the active site because of charge neutrality. Another explanation is that within this experimental design, BSA and IgG may be attracted via different mechanisms to ZS, not the glass.
Figure 6a and b show the same experiments, performed in MPEG coated channels. Since the BSA arrived much later than the IgG in a typical injection, and the intensity was very low, we were not able to perform the injection with both proteins in the same channel. However, even with the individual proteins, we can observe a similar behavior as in the ZS coated channel. As with ZS, the coating seems to be particularly effective in preventing IgG adsorption, while the BSA is barely discernible. The very low fluorescence value of the BSA peak suggests that most of the sample is irreversibly adsorbed to the wall already during the first injection. The shape of the BSA peak over time, with a pronounced tail, suggests that adsorption continue also during subsequent injections, but that this phenomenon is at least partially reversible. Similarly, the IgG peak shows a slight shift and broadening, which indicates that there are small amounts of absorption here as well.
Conclusion
Surface coatings are increasingly common in microfluidics-based applications that focus on biomolecules and protein separations. In this paper, we have compared five commonly used coatings, and compared their performance in terms of successful silanization of channel surfaces, stability over time, and antifouling performance. BSA and IgG have been used as model proteins, given their abundance and adsorption behavior.
Fluorescence measurements of adsorption on glass slides and straight channels confirm that simple hydrophilic (MPEG) and zwitterionic coatings (ZP, ZS) are very effective at preventing adsorption of proteins, whereas hydrophilic coatings (3-MPS, 3-APS) are not. Similarly, measurements of residual fluorescence in straight channels exposed to electrokinetic buffer rinse confirm the superior performance of MPEG, ZP and ZS, as well as the stronger fouling characteristics of IgG compared to BSA. We hypothesize that the poor performance of 3-MPS and 3-APS is due to disulfide linkages with 3-MPS or carboxylate-salt interactions between the protein c-terminus and 3. In addition, flow within microchannels affects adsorption most likely due to a shearing effect at the wall, which can affect MPEG channels adversely.
Finally, given the widespread use of microfluidic separation techniques for identification and characterization of complex samples in a variety of applications, we have compared the behavior of MPEG and ZS during competitive injections of protein mixtures. Although it is not surprising that ZS shows a superior performance compared to MPEG, the very large adsorption of BSA in the MPEG-coated channel is unexpected from the glass slide and channel measurements.
Although ZS showed superior performance in regards to microfluidic separations, it is also important to consider the coating stability. Measurements of zeta-potential over a period of six weeks revealed that zwitterionic coatings tend to degrade over time. In case of ZS, we hypothesize that the behavior observed is the result of an island morphology and multi-layer formation in the coating, due to single point silanization, with concomitant oligomerization. For ZP, we hypothesize that interactions in the presence of sodium borate buffer removed weakly bound silanes via ionic interactions or hydrolyzed the phosphodiester bond of the functional chain, thus shifting charge neutrality and density.
Based on these findings, we conclude that MPEG is the most effective surface coating for applications where stability of the coating over time is critical. However, for separation experiments of BSA- or IgG-like proteins, where the channel is used shortly after coating, silanized ZS has superior anti-fouling characteristics, and should be used for the most accurate measurements. Importantly, our findings also highlight that, when optimizing microfluidic-/surface- based protein assays, sample composition needs to be taken into account, as different proteins may behave differently based on their structure and properties such as molecular weight (BSA: 66.5 kDa, Igg: 150- 170 kDa), isoelectric point, flexibility, as well as maybe distribution of hydrophobic groups.
References
- Pennathur S, Santiago JG (2006) Electrokinetic Transport in Nanochannels. 1. Theory. Anal Chem 78: 972-973.
- Huber DE, Markel ML, Pennathur S, Patel KD (2009) Oligonucleotide Hybridization and Free-solution Electrokinetic Separation in a Nanofluidic Device. Lab Chip 9: 2933-2940.
- Ajdari A, Bocquet L (2006) Giant amplification of interfacially driven transport by hydrodynamic slip: diffusio-osmosis and beyond. Phys Rev Lett 96: 186102.
- van der Heyden FH, Bonthuis DJ, Stein D, Meyer C, Dekker C (2007) Power generation by pressure-driven transport of ions in nanofluidic channels. Nano Lett 7: 1022-1025.
- Pallandre A, de Lambert B, Attia R, Jonas AM, Viovy JL (2006) Surface treatment and characterization: Perspectives to electrophoresis and lab-on-chips. Electrophoresis 27: 584-610.
- Taylor RB (1993) Capillary electrophoresis-priciples, practice and applications: S. F. Y. Li, Elsevier, Amsterdam, 1992. Pages: xxvi + 582. Dfl 395.00. ISBN 0-444-89433-0. Talanta 40: 770.
- Bello MS, Zhukov MY, Righetti PG (1995) Combined effects of non-linear electrophoresis and non-linear chromatography on concentration profiles in capillary electrophoresis. J Chromatogr A 693: 113-130.
- DolnÃk V (2006) Capillary electrophoresis of proteins 2003-2005. Electrophoresis 27: 126-141.
- Gubala V, Siegrist J, Monaghan R, O’Reilly B, Gandhiraman RP, et al. (2013) Simple approach to study biomolecule adsorption in polymeric microfluidic channels. Anal Chim Acta 760: 75-82.
- Ermakov SV, Zhukov MY, Capelli L, Righetti PG (1995) Wall adsorption in capillary electrophoresis. Experimental study and computer simulation. J Chromatogr A 699: 297-313.
- Yoon J-Y, Kim C-J, Garrell RL (2003) Preventing biomolecular adsorption in electrowetting-based biofluidic chips. Anal Chem 75: 5097-5102.
- Kim J (2002) Protein adsorption on polymer particles. J Biomed Mater Res 2: 4373-438.
- Horbett TA, Brash JL (1995) Proteins at Interfaces II Fundamentals and Applications. ACS Symposium series.
- Malmsten M (1998) Formation of Adsorbed Protein Layers. J Colloid Interface Sci 207: 186-199.
- Mrksich M, Whitesides GM (1996) Using self-assembled monolayers to understand the interactions of man-made surfaces with proteins and cells. Annu Rev Biophys Biomol Struct 25: 55-78.
- Ostuni E, Chapman RG, Liang MN, Meluleni G, Pier G, et al. (2001) Self-Assembled Monolayers That Resist the Adsorption of Cells. Langmuir 17: 6336-6343.
- Szleifer I (1997) Polymers and proteins: interactions at interfaces. Curr Opin Solid State Mater Sci 2: 337-344.
- Yuan Y, Oberholzer MR, Lenhoff AM (2000) Size does matter: Electrostatically determined surface coverage trends in protein and colloid adsorption. Colloids Surfaces A Physicochem Eng Asp 165: 125-141.
- Yoon J-Y, Garrell RL (2008) Biomolecular Adsorption in Microfluidics. In: Li D (Ed.) Encyclopedia of Microfluidics and Nanofluidics, Springer pp. 68-76.
- Norde W (1986) Adsorption of proteins from solution at the solid-liquid interface. Adv Colloid Interface Sci 25: 267-340.
- Chen AB, Nashabeh W, Wehr T (2001) CE in Biotechnology: Practical Applications for Protein and Peptide Analyses. Chromatographia CE Series, Springer.
- Lee C, Blanchard W, Wu C (1990) Direct control of the electroosmosis in capillary zone electrophoresis by using an external electric field. Anal Chem 62: 1550-1552.
- Hayes MA, Kheterpal I, Ewing AG (1993) Electroosmotic flow control and surface conductance in capillary zone electrophoresis. Anal Chem 65: 2010-2013.
- Green JS, Jorgenson W (1989) Minimizing Adsorption Of Proteins On Fused Silica In Capillary Zone Electrophoresis By The Addition Of Alkali Metal Salts To The Buffers. J Chromatogr 478: 63-70.
- Bushey MM, Jorgenson JW (1989) Capillary electrophoresis of proteins in buffers containing high concentrations of zwitterionic salts. J Chromatogr 480: 301-310.
- Lauer HH, McManigill D (1986) Capillary zone electrophoresis of proteins in untreated fused silica tubing. Anal Chem 58: 166-170.
- Belder D, Ludwig M (2003) Surface modification in microchip electrophoresis. Electrophoresis 24: 3595-3606.
- Hjertén S (1985) High-performance electrophoresis Elimination of electroendosmosis and solute adsorption. J Chromatogr A 347: 191-198.
- Nashabeh W, El Rassi Z (1991) Capillary zone electrophoresis of proteins with hydrophilic fused-silica capillaries. J Chromatogr 559: 367-383.
- Bruin GJM, Kuhlman RH, Zegers K, Kraak JC, Poppe H (1989) Capillary Zone Electrophoretic Separations Of Proteins In Polyethylene Glycol-Modified Capillaries. J Chromatogr 47: 429-436.
- van Reenen A, de Jong AM, den Toonder JMJ, Prins MWJ (2014) Integrated lab-on-chip biosensing systems based on magnetic particle actuation - a comprehensive review. Lab Chip 14: 1966-1986.
- Milanova D, Chambers RD, Bahga SS, Santiago JG (2012) Effect of PVP on the electroosmotic mobility of wet-etched glass microchannels. Electrophoresis 33: 3259-3262.
- Jiang Y, Wang H, Li S, Wen W (2014) Applications of micro/nanoparticles in microfluidic sensors: A review. Sensors 14: 6952-6964.
- Banerjee I, Pangule RC, Kane RS (2011) Antifouling coatings: Recent developments in the design of surfaces that prevent fouling by proteins, bacteria, and marine organisms. Adv Mater 23: 690-718.
- Riche CT, Zhang C, Gupta M, Malmstadt N (2014) Fluoropolymer surface coatings to control droplets in microfluidic devices. Lab Chip 14: 1834-1841.
- Sapsford KE, Ligler FS (2004) Real-time analysis of protein adsorption to a variety of thin films. Biosens Bioelectron 19: 1045-1055.
- Luk YY, Kato M, Mrksich M (2000) Self-assembled monolayers of alkanethiolates presenting mannitol groups are inert to protein adsorption and cell attachment. Langmuir 16: 9604-9608.
- Horvath J, DolnÃk V (2001) Polymer wall coatings for capillary electrophoresis. Electrophoresis 22: 644-655.
- Doherty EA, Meagher RJ, Albarghouthi MN, Barron AE (2003) Microchannel wall coatings for protein separations by capillary and chip electrophoresis. Electrophoresis 24: 34-54.
- Wong I, Ho CM (2009) Surface molecular property modifications for poly(dimethylsiloxane) (PDMS) based microfluidic devices. Microfluid Nanofluidics 7: 291-306.
- Estephan ZG, Jaber JA, Schlenoff JB (2010) Zwitterion-stabilized silica nanoparticles: toward nonstick nano. Langmuir 26: 16884-16889.
- Wu L, Guo Z, Meng S, Zhong W, Du Q, et al. (2010) Synthesis of a zwitterionic silane and its application in the surface modification of silicon-based material surfaces for improved hemocompatibility. ACS Appl Mater Interfaces 2: 2781-2788.
- Pennathur S, Crisalli P (2013) Low Temperature Fabrication and Surface Modification Methods for Fused Silica Micro- and Nanochannels. MRS Proc 1659: 15-26.
- Estephan ZG, Schlenoff PS, Schlenoff JB (2011) Zwitteration as an alternative to PEGylation. Langmuir 27: 6794-6800.
- Pennathur S, Santiago JG (2005) Electrokinetic transport in nanochannels. 2. Experiments. Anal Chem 77: 6782-6789.
- Sze A, Erickson D, Ren L, Li D (2003) Zeta-potential measurement using the Smoluchowski equation and the slope of the current-time relationship in electroosmotic flow. J Colloid Interface Sci 261: 402-410.
- Bharadwaj R, Santiago JG, Mohammadi B (2002) Design and optimization of on-chip capillary electrophoresis. Electrophoresis 23: 2729-2744.
- Liu H, May K (2012) Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs 4: 17-23.
- Smith EA, Chen W (2008) How to prevent the loss of surface functionality derived from aminosilanes. Langmuir 24: 12405-12409.
- Li L, Chen S, Zheng J, Ratner BD, Jiang S (2005) Protein Adsorption on Oligo (ethylene glycol) -Terminated Alkanethiolate Self-Assembled Monolayers?: The Molecular Basis for Nonfouling Behavior. J Phys Chem B 109: 2934-2941.
- Chen S, Li L, Zhao C, Zheng J (2010) Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 5: 5283-5293.
- McNaught AD, Wilkinson A (1997) Compendium of Chemical Terminology. Oxford: Blackwell Scientific Publications.
- Kirby BJ (2010) Micro- and Nanoscale Fluid Mechanics: Transport in Microfluidic Devices. Cambridge University Press.
- Andersen MB, Bruus H, Bardhan JP, Pennathur S (2011) Streaming current and wall dissolution over 48 h in silica nanochannels. J Colloid Interface Sci 360: 262-271.
- Jensen KL, Kristensen JT, Crumrine AM, Andersen MB, Bruus H, et al. (2011) Hydronium-dominated ion transport in carbon-dioxide-saturated electrolytes at low salt concentrations in nanochannels. Phys Rev E 83: 056307.
- Yang W, Zhang L, Wang S, White AD, Jiang S (2009) Functionalizable and ultra-stable nanoparticles coated with zwitterionic poly(carboxybetaine) in undiluted blood serum. Biomaterials 30: 5617-5621.
- Mathé C, Devineau S, Aude JC, Lagniel G, Chédin S, et al. (2013) Structural determinants for protein adsorption/non-adsorption to silica surface. PLoS One 8: e81346.
- Vogler EA (2012) Protein adsorption in three dimensions. Biomaterials 33: 1201-1237.
- Hlady V, Buijs J, Jennissen HP (1999) Methods for studying protein adsorption. Methods Enzymol 309: 402-429.
- Mathes J, Friess W (2011) Influence of pH and ionic strength on IgG adsorption to vials. Eur J Pharm Biopharm 78: 239-247.
- Lionello A, Josserand J, Jensen H, Girault HH (2005) Protein adsorption in static microsystems: effect of the surface to volume ratio. Lab Chip 5: 254-260.
- Chen S, Zheng J, Li L, Jiang S (2005) Strong resistance of phosphorylcholine self-assembled monolayers to protein adsorption: Insights into nonfouling properties of zwitterionic materials. J Am Chem Soc 127: 14473-14478.
- Schoch RB, Renaud P (2008) Transport phenomena in nanofluidics. Rev Mod Phys 80: 839-883.
- Karnik R, Castelino K, Majumdar A (2006) Field-effect control of protein transport in a nanofluidic transistor circuit. Appl Phys Lett 88: 1-3.
- Volkmuth WD, Duke T, Wu MC, Austin RH, Szabo A (1994) DNA electrodiffusion in a 2D array of posts. Phys Rev Lett 72: 2117-2120.
- Lee JH, Chung S, Kim SJ, Han J (2007) Poly(dimethylsiloxane)-based protein preconcentration using a nanogap generated by junction gap breakdown. Anal Chem 79: 6868-6873.
- Wang Y-C, Choi MH, Han J (2004) Two-dimensional protein separation with advanced sample and buffer isolation using microfluidic valves. Anal Chem 76: 4426-4431.
- Kaji N, Tezuka Y, Takamura Y, Ueda M, Nishimoto T, et al. (2004) Separation of long DNA molecules by quartz nanopillar chips under a direct current electric field. Anal Chem 76: 15-22.
- Doyle PS, Bibette J, Bancaud A, Viovy JL (2002) Self-assembled magnetic matrices for DNA separation chips. Science 295: 2237.
- Tegenfeldt JO, Prinz C, Cao H, Huang RL, Austin RH, et al. (2004) Micro- and nanofluidics for DNA analysis. Anal Bioanal Chem 378: 1678-1692.
- Lutanie E, Voegel JC, Schaaf P, Freund M, Cazenave JP, et al. (1992) Competitive adsorption of human immunoglobulin G and albumin: consequences for structure and reactivity of the adsorbed layer. Proc Natl. Acad Sci USA 89: 9890-9894.
Citation: Williams S, Venkateswaran N, O’Donnell TDB, Crisalli P, Helmy S, et al. (2016) Assessing Stability, Durability, and Protein Adsorption Behavior of Hydrophilic Silane Coatings in Glass Microchannels. J Anal Bioanal Tech 7:318. DOI: 10.4172/2155-9872.1000318
Copyright: © 2016 Williams S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Open Access Journals
Article Tools
Article Usage
- Total views: 15302
- [From(publication date): 6-2016 - Aug 23, 2025]
- Breakdown by view type
- HTML page views: 14206
- PDF downloads: 1096