Research Article Open Access
High Throughput LC-MS/MS Method for Simultaneous Estimation of 9-Cis-Retinoic Acid and its Metabolite 4-Oxo-9-Cis-Retinoic Acid in Human Plasma and its Application to a Bioequivalence Study
Arabinda Saha*, Poonam Vats, Richa Thakur, Arshad Khuroo and Tausif MonifDepartment of Clinical Pharmacology and Pharmacokinetics, Sun Pharmaceutical Industries Ltd., Haryana, India
- *Corresponding Author:
- Arabinda Saha
Department of Clinical Pharmacology and Pharmacokinetics
Sun Pharmaceutical Industries Ltd., GP-5, HSIDC
Old Delhi Gurgaon Road, Udyog Vihar Industrial Area
Gurgaon-122 015, Haryana, India
Tel: +91-22-43244324
E-mail: arabinda.saha@sunpharma.com
Received date: October 07, 2015; Accepted date: October 26, 2015; Published date: November 02, 2015
Citation: Saha A, Vats P, Thakur R, Khuroo A, Monif T (2015) High Throughput LC-MS/MS Method for Simultaneous Estimation of 9-Cis-Retinoic Acid and its Metabolite 4-Oxo-9-Cis-Retinoic Acid in Human Plasma and its Application to a Bioequivalence Study. J Anal Bioanal Tech S13:001. doi:10.4172/2155-9872.S13-001
Copyright: © 2015 Saha A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Analytical & Bioanalytical Techniques
Abstract
A liquid chromatography mass-spectrometric method is described for the simultaneous estimation of 9-cisretinoic acid and 4-oxo-9-cis-retinoic acid in human plasma using 13-cis-retinoic acid-d5 as an internal standard. Chromatographic separation of these retinoids was achieved on Chromolith Performance RP18 (100 × 4.6 mm) column using binary flow program of two mobile phase. 9-cis-retinoic acid and 4-oxo-9-cis-retinoic acid were extracted from human plasma by liquid-liquid extraction with a mixture of ethyl acetate, n-hexane and iso propyl alcohol (30:65:5, v/v/v). Quantification was achieved by monitoring transitions of m/z 299.4→255.2 for 9-cisretinoic acid, m/z 313.4→269.3 for 4-oxo-9-cis-retinoic acid and 304.4→260.2 for isotretinoin-d5 in multiple reaction monitoring, using turbo ion source in negative polarity. The linearity was established over a dynamic range of 2.85- 450.51 ng/mL and 2.33-180.57 ng/mL for 9-cis-retinoic acid and 4-oxo-9-cis-retinoic acid, respectively. At the lower limit of quantification level, the intra-and inter-day precision values of 9-cis-retinoic acid were within 5.9% and 6.0%, respectively, whereas that of 4-oxo-9-cis-retinoic acid were within 13.8% and 11.3%, respectively. The method was successfully applied for the simultaneous estimation of 9-cis-retinoic acid and 4-oxo-9-cis-retinoic acid in healthy volunteers, after oral administration of 30 mg alitretinoin capsule for bioequivalence study.
Keywords
9-cis-retinoic acid; 4-oxo-9-cis-retinoic acid; Bioequivalence study; Geometrical isomer; LC-MS/MS; Retinoid
Introduction
Retinoids are a class of naturally occurring and synthetic compounds that are structurally related to retinol (vitamin A) and play a significant role in a variety of physiological processes, i.e., vision, morphogenesis, growth and differentiation of tissues, reproduction, skin disorders and immune modulation [1-3]. Alitretinoin (9-cis-retinoic acid, 9-cRA), isotretinoin (13-cisretinoic acid, 13-cRA), tretinoin (all-trans-retinoic acid, aTRA) are endogenously occurring vitamin A derivatives and geometrical isomer to each other. 9-cRA have recently become the only licensed systemic agent for severe chronic hand eczema unresponsive to treatment with potent topical corticosteroids [4]. The two families of retinoid nuclear receptors that have been identified, the retinoic acid (RA) receptors [5,6] and the retinoid X receptors [7,8], have ligand-binding domains which share only 29% homology.
The RA receptors (α, β and γ) bind the naturally occurring retinoid aTRA with high affinity, whereas the retinoid X receptors (α, β and γ) are activated by but do not bind aTRA [9]. This multiplicity of receptors and gene pathways may, in part, explain the diverse effects of retinoids on a wide range of cellular processes. 9-cRA is capable of binding and transactivating both retinoid X receptors as well as RA receptors [5,10,11]. This may, in part, account for its enhanced potency, compared with aTRA, in inhibiting the growth and inducing the differentiation of a spectrum of human tumors in vitro [1,12,13]. Isomerization is an important metabolic pathway of RAs, since it results in metabolites having different effects due to diverse mechanism of action. Another step in RA metabolism is the oxidation, wherein RAs get converted to their respective 4-oxo-metabolites by cytochrome P450, which is the main metabolic pathway after the application of pharmacological doses. At higher concentrations, 4-oxo-9-cis-retinoic acid (4-oxo-9-cRA) show the same effects as 9-cRA on in vitro cell proliferation and differentiation [14].
The total estimated concentration of RAs in human plasma is ~ 4.9 ng/mL and aTRA pr esent 75% of the estimated concentration [15]. The residual concentration (~ 1.2 ng/mL) is possible due to the presence of other RAs (13-cRA, 9-cRA, retinol etc.). As per our present investigation the maximum concentration of 9-cRA in human plasma was found to be 0.3 ng/mL, the same being also reported by Lanvers et al. [16]. Therefore, for the accurate estimation of 9-cRA in human plasma and reliable characterization of pharmacokinetic profile, it is essential to develop a sensitive method with low LOQ (<3.0 ng/mL) for 30 mg alitretinoin capsule. In order to develop a method with the desired LOQ, it was necessary to use MS-MS detection, owing to the advantages it holds.
Several HPLC methods have been developed by Lanvers et al. [16], Disdier et al. [17], Miyagi et al. [18], Ruhl et al. [19], Dzerk et al. [20], Schmidt et al. [21] for simultaneous estimation of 9-cRA and 4-oxo- 9-cRA in human and animal biological matrices. But these reported methods, sample preparation is more critical and also require lengthy analysis time (>40.0 min) for separation of geometrical isomers. Marchetti et al. [22] estimated the concentration of 9-cRA in rabbit plasma within 12.5 min run time by using HPLC system. Arnold et al. [23] developed UHPLC-MS method for estimation of multiple retinoids in human serum, by which the estimation of 9-cRA is feasible within 15.0 min run time. However, the major metabolite of 9-cRA (i.e., 4-oxo-9-cRA) was neither estimated nor seperated from its geometrical isomers by Marchetti et al. [22], Arnold et al. [23]. Wang et al. [24] developed LC-MS/MS method for simultaneous estimation of 9-cRA, 13-cRA and aTRA in rat prostrate, but the method had lengthy analysis run time (>27.0 min) and evaluation of co-elute matrix effect was not addressed. The limit of quantification of RA was improved by Napoli [15], using gas chromatography - mass spectrometry (GC-MS). However, GC-MS requires derivatization of samples before analysis, which complicates and prolongs sample preparation. Additionaly, the high temperature (typically 230°C) used during GC separation might cause on-column isomerisation and oxidation of RA, which might lead to psedo estimation of RA in matrix.
Therefore, while developing method for the simultaneous estimation of 9-cRA and 4-oxo-9-cRA, above mentioned limitations were taken into consideration. Method presented in this article has number of advantages over the already reported methods, some of which are: lesser aliquot volume (100 μL) compared to those used by Lanvers et al. [16], Dzerk et al. [20], Disdier et al. [17], and shortest analysis time (18.0 min) till date for simultaneous estimation of 9-cRA and 4-oxo-9- cRA in human plasma. Furthermore, a systematic evaluation of matrix interference was investigated by employing liquid-liquid extraction technique, in order to efficiently nullify the level of matrix effect and detailed stability studies were carried out in both types of human matrix (i.e., plasma and whole blood) as per current regulatory guidelines. The current method highlights selectivity of 9-cRA and 4-oxo-9-cRA in different human plasma (normal, haemolyzed and lipemic plasma). The method was successfully used to evaluate the bioequivalence of the pharmaceutical formulations of alitretinoin capsule in healthy Indian male volunteers.
Experimental
Chemicals and materials
Alitretinoin (9-cis-retinoic acid, 9-cRA), isotretinoin (13-cisretinoic acid, 13-cRA), tretinoin (all-trans-retinoic acid, aTRA), 4-oxo-alitretinoin (4-oxo-9-cis-retinoic acid, 4-oxo-9-cRA), 4-oxoisotretinoin (4-oxo-13-cis-retinoic acid, 4-oxo-13-cRA), 4-oxotretinoin (4-oxo-all-trans-retinoic acid, 4-oxo-aTRA) and 4-oxo- 9,13-dicis retinoic acid (4-oxo-9,13-cRA) were procured from Ranbaxy Research Laboratories. Isotretinin-d5 (13-cis-retinoic acid-d5, 13-cRA-d5) was procured from Toronto Research Chemicals Inc., Canada respectively. Ammonium acetate, acetonitrile, dimethyl sulfoxide (DMSO) and methanol were procured from Fluka (Sigma- Aldrich, steinheim, USA). Glacial acetic acid, iso propyl alcohol (IPA) and liquor ammonia were obtained from Fischer Scientific, India. Ethyl acetate and n-hexane were procured from SD fine chemicals, India. Milli-Q water (Millipore, Moscheim Cedex, France) was used in the preparation of solutions. Human plasma lots containing K3EDTA (Ethylene diamine tetra acetic acid tri potassium salt, as anticoagulant) were obtained from Biological Specialty Corporation, PA.
Laboratory precautions
Since RAs are sensitive to light, all experiments (Sample preparation and LC-MS/MS analysis) were carried out in dark rooms under red light condition to prevent photoisomerization and photodegradation. Due to standardized LC-MS/MS analysis and sample preparation all laboratory work was performed at 20°C.
LC-MS/MS instrumentation and chromatographic conditions
Chromatographic separation was carried out on a Shimadzu scientific instrument (Shimadzu Corporation; Kyoto, Japan) with Merck Chromolith Performance RP18 (100 × 4.6 mm) column at 45°C, using mobile phase-A, containing 5 mM ammonium acetate buffer (pH 3.0)-methanol (35:65, v/v) and mobile phase-B, containing 5 mM ammonium acetate buffer (pH 3.0)-methanol-acetonitrile (30:30:40, v/v/v). Mobile phase-A was delivered at a flow rate of 1.8 mL/min from 0.00 to 6.20 min and 17.01 to 18.00 min and mobile phase-B was delivered at a flow rate of 1.8 mL/min from 6.21 min to 17.00 min. The total analysis time for each sample was 18.0 min. The ionization and detection were carried out on a triple quadruple mass spectrometer, MDS Sciex API-4000 (Sciex Division of MDS, Toronto, Ontario, Canada), equipped with electrospray ionization operated in negative polarity using multiple reaction monitoring (-MRM).
The parameters, optimized by infusing solution of 9-cRA, 4-oxo- 9-cRA and 13-cRA-d5 into the mass spectrometer were as follows: collision activated dissociation (CAD), curtain gas (CUR), nebulizer gas (GS1), heater gas (GS2), 12, 25, 65 and 40 psi, respectively; ion spray voltage, -4500 V, ion source temperature, 650°C; declustering potential (DP), entrance potential (EP), collision energy (CE), and collision cell exit potential (CXP) were optimized and set at -48, -10, -22, -6 V, respectively, for all compounds. Both quadrupole 1 and quadrupole 3 were maintained at unit resolution, and the dwell time was set at 400 millisec for all compounds. The mass transitions were selected as 299.4→255.2 for 9-cRA, 313.4→269.3 for 4-oxo-9-cRA and 304.4→260.2 for 13-cRA-d5. The data acquisition and processing were performed by Analyst version 1.4.2 software (MDS Sciex, Toronto, Canada). For quantification, the peak area ratios of the target ions of the analyte to those of the internal standard were compared with weighted 1/X2 (Where, X=Drug concentration) least squares calibration curves in which the peak area ratios of the calibration standards were plotted versus their concentrations.
Preparation of stock solutions, calibration standards and quality control samples
Two separate stock solutions of 9-cRA and 4-oxo-9-cRA were prepared for bulk spiking of calibration standards (CS) and quality control (QC) samples for method validation exercises as well as clinical sample analysis. Stock solutions of 9-cRA, 4-oxo-9-cRA and 13-cRA-d5 (chemical structures are shown in Figure 1) were prepared in DMSO:IPA (50:50, v/v) at concentration of 1 mg/mL and stored below -15°C. Separate working solutions of 9-cRA and 4-oxo-9-cRA were prepared for preparation of CS and QC samples by appropriate dilution in acetonitrile-water (50:50, v/v). Blank human K3EDTA plasma was screened prior to spiking to ensure that there is no significant endogenous interference at the retention time (RT) of 9-cRA, 4-oxo-9-cRA and 13-cRA-d5. Eight non zero CS at nominal values of 2.85, 8.06, 16.12, 36.63, 83.25, 189.21, 378.43 and 450.51 ng/ mL concentrations were used for spiking 9-cRA. Similar calibrators at nominal values of 2.33, 6.66, 13.32, 23.78, 42.47, 75.84, 151.68 and 180.57 ng/mL for 4-oxo-9-cRA were prepared together with 9-cRA, so that individual spiking of analyte was not required. Four in-house QC samples represented at lower limit of quantification (LOQQC), low quality control (LQC), medium quality control (MQC) and high quality control (HQC) at respective nominal values of 2.87, 8.14, 189.29 and 378.57 ng/mL for 9-cRA and 2.34, 6.68, 75.94 and 151.87 ng/mL for 4-oxo-9-cRA were prepared. Spiking was carried out in ice cold water bath, under red light condition. The bulk spiked CS and QC samples were aliquoted in amber color polypropylene tubes and were stored below -50°C and protected from light until further use. The working solution of 13-cRA-d5 (400.0 ng/mL) for routine use, was prepared by diluting the stock solution of 13-cRA-d5 in acetonitrile-water (50:50, v/v) and kept in ice cold water bath, under red light condition until use.

Figure 1: Chemical structure. (A) Alitretinoin (9-cis-retinoic acid; 9-cRA), (B) 4-oxo-alitretinoin (4-oxo-9-cis-retinoic acid; 4-oxo-9-cRA) and (C) Isotretinoin-d5 (13-cis-retinoic acid-d5; 13-cRA-d5).
Sample preparation
Before analysis, the calibration standards, QC samples and clinical samples were thawed in ice cold water bath, under red light condition. Plasma sample (100 μL) was aliquoted in amber color polypropylene tube (16 × 125 mm) and 50 μL of the internal standard working solution (400.0 ng/mL) was added and 300 μL of 5 mM ammonium acetate buffer (pH 6.2) was added and vortexed, followed by the addition of 4 mL of an extraction mixture, containing ethyl acetate, n-hexane and IPA (30:65:5, v/v/v) was added. The sample was extracted on a reciprocating shaker at 100 rpm for 15 min. After centrifugation at 4000 rpm for 5 min, the aqueous layer was frozen in a dry ice-methanol bath. The organic layer was decanted into a glass tube (13 × 100 mm) and evaporated to dryness under nitrogen stream at 40°C. The dried residue was reconstituted with 300 μL of reconstitution solution, containing 5 mM ammonium acetate buffer (pH 9.0)-methanolacetonitrile (30:30:40, v/v/v) and 25 μL of the reconstituted sample was injected into LC-MS/MS system for analysis.
Method validation
A thorough and complete method validation of 9-cRA and 4-oxo- 9-cRA in human K3EDTA plasma was carried out, as per the United States Food and Drug Administration [25] and European Medicines Agency [26]. The method was validated for selectivity, sensitivity, linearity, precision, accuracy, process efficiency, dilution integrity, matrix effect, re-injection reproducibility and stability of 9-cRA and 4-oxo-9-cRA during both short term sample processing and long term storage.
Selectivity and signal-to-noise (S/N) ratio
The selectivity of the method towards endogenous plasma matrix components and concomitant medications was assessed after screening ten lots (6 normal, 2 haemolyzed and 2 lipemic plasma) of human K3EDTA plasma, free from all analyte of interest. These samples were processed using the proposed extraction protocol and analyzed with the set chromatographic conditions of both analyte at lower limit of quantification (LOQ) level. The peak area of the co-eluting components or interferences in blank sample at the RT of 9-cRA, 4-oxo-9-cRA and 13-cRA-d5 should be <20% of mean peak area of analyte (9-cRA and 4-oxo-9-cRA) and <5% of mean peak area of 13-cRA-d5 in spiked LOQ sample. The sensitivity was demonstrated by determining the signal to noise (S/N) ratio in all ten lots of screened plasma and spiked LOQ samples. The S/N ratio of spiked LOQ samples was calculated by using following formula:
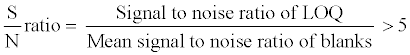
Linearity
Three calibration curves were used to demonstrate the linearity of the method. The ratio of area responses for analyte was used for regression analysis. Each calibration curve was analyzed individually by using least square weighted (1/X2) linear regression (obtained by best fit method). Back-calculations were made from these curves to determine the concentration of 9-cRA and 4-oxo-9-cRA in each calibrator. The correlation coefficient, r>0.99 was mandatory for all the calibration curves to be accepted. Intra- and inter-day precision and accuracy were determined on the basis of six replicates of QC samples included in each run. The lowest standard concentration on the calibration curve was to be accepted as LOQ, if the analyte response was at least five times more than that of drug-free (blank) extracted plasma. In addition, the analyte peak of LOQ sample should be identifiable, discrete, and reproducible with accuracy within ± 20% and a precision ≤ 20%. The deviation of standards other than LOQ from the nominal concentration should not be more than ± 15%.
Precision and accuracy
The precision of the method was determined by calculating the % CV at each QC level. The deviation at each concentration level from the nominal concentration should be within ± 15%, excluding at LOQQC level (± 20%). Similarly, the mean accuracy should not deviate ± 15%, excluding at LOQQC level (± 20%).
Process efficiency and matrix effect
The process efficiency (PE) of 9-cRA and 4-oxo-9-cRA in QC samples (LQC, MQC and HQC level) was determined by comparing the mean peak area response of analyte in six replicates of extracted QC samples (spiked before extraction) with those of unextracted sample (neat sample) containing analyte at concentrations equivalent to those obtained in the final extracted concentration for analyte in the extracted QC samples. The PE of internal standard (13-cRA-d5) was determined at the working concentration (400.0 ng/mL) in a similar way. Process efficiency (PE) of analytes and internal standard were estimated by using the following equation:
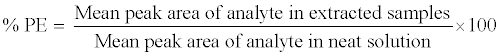
The absolute matrix effect (AME) was estimated by the following equation:
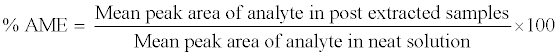
Where, AME=1 indicates no matrix effect, AME<1 indicates ionsuppression and AME>1 indicates ion-enhancement. As extraction protocol involves a terminal drying step, hence spiking (addition of reference sample) was carried out in post-extracted blank plasma sample to perform AME. The concentration of both analyte and internal standard in reference sample representing the final extrcated concentrations in QC samples (at LQC, MQC and HQC level). The control sample was a reference solution prepared at an appropriate concentration in reconstitution solution.
Relative Matrix effect (RME) was evaluated by using six lots of human K3EDTA plasma including one hemolyzed and one lipemic plasma lot. In each lot, aqueous dilutions of 9-cRA and 4-oxo-9-cRA were spiked at LOQQC and HQC level and processed in duplicate samples and the area ratio (i.e., peak area of analyte/peak area of internal standard) was used to check the acceptability of the result. The standard deviation for each lot was calculated, along with %CV and % bias at each level. The deviation of the standards should not be more than ± 15% of their respective nominal concentration and at least 90% of the lots at each QC level should be within the aforementioned criteria.
Stability excercises
Stability experiments were carried out to examine the stability of 9-cRA and 4-oxo-9-cRA in stock solution and in plasma samples under different conditions. Stock solution stability was performed by comparing peak area response of 9-cRA, 4-oxo-9-cRA and 13-cRA-d5 in stability sample, with the peak area response of sample prepared from fresh stock solution. Stability studies in plasma were performed at LQC and HQC level using four replicates at each level. The analyte was considered stable if the % change is less than 15, as per US FDA/EMEA guidelines and was calculated by using the following formula:
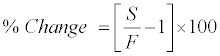
Where, S=Mean concentration of stability samples and F=Mean concentration of freshly spiked samples.
The bench top stability was determined by storing spiked QC samples for ~ 6.3 h in ice cold water bath, under red light condition before processing. The auto sampler stability was determined by storing reconstituted QC samples for ~ 128 h in auto sampler (at 5°C) before being analyzed. The freeze-thaw stability was conducted by comparing the stability samples that had been frozen at -50°C and thawed in ice cold water bath three times under red light condition, with freshly spiked QC samples. For long term stability evaluation, the concentrations of QC samples obtained after 183 days (below -50°C) were compared with the initial concentrations. Four aliquots each of LQC and HQC concentration level were used for the stability evaluation. All stability exercises were performed against freshly spiked CS.
Human K3EDTA whole blood spiked with working solutions (at LQC and HQC level) were prepared and after spiking spiked sample was split into two aliquots (A and B). Aliquot A was placed for 10 min in ice cold water bath, centrifuged at 4°C and the resulting plasma was used as comparison sample. Aliquot B was kept in ice cold water bath and under red light condition for 2 h, centrifuged at 4°C and the resulting plasma (stability samples) was analyzed with the comparison samples in the same batch to access the % stability during the sample collection process. The analytes were considered stable if the % stability was within 85-115. The % stability was calculated by using following formula:
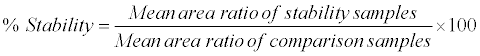
Re-injection reproducibility and dilution integrity
Re-injection reproducibility was performed by re-injecting all QC samples (i.e., LOQQC, LQC, MQC and HQC) from an accepted precision-accuracy batch during method validation. The calculated concentration of re-injected QC samples was determined against the CS samples from the same precision and accuracy batch which was analyzed 48 h before. The % difference between original and re-injected value was calculated by using following formula:
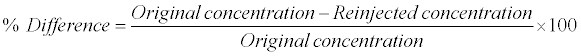
The dilution integrity experiment was performed with an aim to validate the dilution test to be carried out on higher analyte concentrations above upper limit of quantification (ULOQ), which may be encountered during real clinical sample analysis. Dilution integrity test was performed by preparing samples at a concentration approximately two times the concentration of 90% ULOQ. These samples were diluted to two and four times with blank plasma to bring the concentration within calibration curve and then analyzed against fresh CS samples. The acceptance criteria for the diluted QC samples are the same as that of QC samples in precision and accuracy run.
Carry over test
Carry over was assessed following injection of a blank plasma sample immediately after three repeats of the Upper Limit Of Quantification (ULOQ) and the peak area response was checked.
Pharmacokinetic study
The method was applied to an open-label, balanced, randomized, three-treatment, three-sequence three-period, single dose, crossover design study of alitretinoin in healthy male volunteers under fasting condition for the assessment of bioequivalence. A single oral dose of alitretinoin 30 mg capsule of Ranbaxy and Toctino 30 mg capsule of Basilea Medical Ltd., UK was given to the human volunteers during the study. The bio-study was carried out in accordance with the principles of Good Clinical Practices defined in the ethical guidelines for Biomedical Research on human participants issued by Indian Council of Medical Research, New Delhi, the ICH E6 Guidance for ‘Guidance on Good Clinical Practice’ and the principles enunciated in the Declaration of Helsinki on 15 healthy volunteers participated in each study from whom prior informed consent was taken. The bio-study protocol was approved by the Jamia Hamdard Institutional Review Board, New Delhi, India. Blood samples were collected before (predose) and at 0.250, 0.500, 0.750, 1.000, 1.333, 1.667, 2.000, 2.333, 2.667, 3.000, 3.500, 4.000, 4.500, 5.000, 6.000, 8.000, 12.000, 16.000, 24.000, 36.000 and 48.000 h post-dose in each period. All blood samples were collected in K3EDTA vacutainers and processed by centrifugation to collect plasma in amber color polypropylene tubes and stored below -50°C until analysis.
Results and Discussion
Optimization of mass spectrometer parameter
Initially mass parameters were tuned in Atmospheric Pressure Chemical Ionization (APCI) and Electrospray Ionization (ESI) ion sources, but inadequate response was observed in APCI ion source. As RA have a carboxylic acid moiety, electrospray ionization in positive polarity was less sensitive than the negative polarity. Negative mode ESI mass spectrum of 9-cRA, 4-oxo-9-cRA and 13-cRA-d5 was dominant with deprotonated (M-H)- ion of m/z 299.4, 313.4 and 304.4 for 9-cRA, 4-oxo-9-cRA and 13-cRA-d5, respectively. Addition of acid further enhanced the intensity of these ions to obtain deprotonated precursor ion peaks at m/z 255.2, 269.3 and 260.2 for 9-cRA, 4-oxo-9-cRA and 13-cRA-d5, respectively. During mass parameter optimization it was observed that CE and CAD are most critical parameters to achieve highest sensitivity with stable response for 9-cRA and 4-oxo-9-cRA.
Chromatographic separation
Photodegradation and geometric (cis/trans) isomerization of RA have been reported [17]. If this isomeric conversion and photodegradation is not inhibited during sample handling condition and chromatographic separation of RA from its geometric isomer is not achieved during analysis, these could lead to pseudo estimation of RA in human plasma.
Initially chromatographic separation of geometrical isomers was carried out by analyzing a pure aqueous mixture, containing all polar RAs (4-oxo-13-cRA, 4-oxo-9-cRA, 4-oxo-aTRA and 4-oxo-9,13-dicis- RA) and non-polar RAs (9-cRA, 13-cRA and aTRA) with isocratic flow of mobile phase. In isocratic condition, polar RAs were eluted early from the column, while the RTs of non-polar RAs were too long (>45.0 min). Thus, two separate dilutions (one containing polar RAs and other containing non-polar RAs) were prepared and individually analyzed for better understanding of the chromatographic separation of these geometrical isomers. The use of volatile buffers like ammonium formate and ammonium acetate (in combination of methanol-acetonitrile) and the pH of buffer solution for chromatographic separation of geometrical isomers has been evaluated. Use of ammonium acetate as a volatile buffer in mobile phase increased the sensitivity of the method by 20 folds as compared to ammonium formate buffer.
During optimization it was observed that, pH 3.0 was appropriate for separation of these geometrical isomers. However, the presence of acetonitrile in mobile phase, resulted in merging of 4-oxo-9-cRA peak with other geometrical isomers. When methanol was used as an organic phase in mobile phase, the polar RAs were well separated, but nonpolar RAs were eluted at high retention time. After re-optimization of the mobile phase composition, it was observed that the combination of acetonitrile-methanol was suitabile for early elution of non-polar RAs from analytical column. Finally, two different mobile phase were employed for seperation of all isomers by using binary flow program (Table 1). By using mobile phase-A, containing 5 mM ammonium acetate buffer (pH 3.0)-methanol (35:65, v/v) polar RAs were separated and with mobile phase-B, containing 5 mM ammonium acetate buffer (pH 3.0)-methanol-acetonitrile (30:30:40, v/v/v) non-polar RAs were seperated. Re-equilibration with 100% mobile phase-A for 1.0 min with same flow rate led to reproducible RT for 4-oxo-RAs in an uninterrupted run.
| Time | % of Mobile phase-A | % of Mobile phase-B | Total flow rate (mL/min) |
|---|---|---|---|
| 0.00 | 100.0 | 0.0 | 1.8 |
| 6.20 | 100.0 | 0.0 | 1.8 |
| 6.21 | 0.0 | 100.0 | 1.8 |
| 17.00 | 0.0 | 100.0 | 1.8 |
| 17.01 | 100.0 | 0.0 | 1.8 |
| 18.00 | 100.0 | 0.0 | 1.8 |
Table 1: Binary flow program.
To achieve the separation of polar and non-polar RAs, numerous C18 and C8 columns were evaluated with different mobile phases. Significant differences in the separation capacity between the different columns was observed and in most columns the separation of 9-cRA and aTRA was not achieved, whereas separation between 13-cRA and aTRA was obtained in virtually all columns tested. By using the chromolith performance RP18 column, baseline separation of aTRA, 9-cRA and 13-cRA was achieved with short analysis time and the resolution among polar RAs was satisfactory. An endogenous matrix peak (vitamin A), which was eluted at the retention time of 11.5 min was well separated from the analyte of interest (9-cRA). 4-oxo-9-cRA peak was well separated from the endogenous matrix peak, which was eluted at retention time of 1.1 min and is the characteristic peak of spiked sample for the mass chromatogram of 4-oxo-RAs.
As shown in Figure 2, the optimized chromatographic conditions enabled separation of all polar and non-polar isomers. After analysis of individual dilution of each isomer, it was confirmed that, the compounds eluted at the RT of 13.2 min, 14.2 min, 15.0 min, 3.5 min, 4.9 min, 6.0 min and 2.6 min were 13-cRA, 9-cRA, aTRA, 4-oxo-13-cRA, 4-oxo-9- cRA, 4-oxo-aTRA and 4-oxo-9,13-dicis-RA, respectively. The LC-MS chromatogram obtained, after analysis of an pure aqueous mixture, containing 9-cRA, 4-oxo-9-cRA and 13-cRA-d5 is shown in Figure 3. The RT for all RAs and their metabolites were fully reproducible on a within-day and day-to-day basis analysis. The coefficients of variation were from 0.1 to 0.3% and 0.2 to 0.8% for within-day and day-to-day analysis, respectively.

Figure 2: LC-MS chromatograms showing separations of polar and non-polar RAs using optimized chromatographic conditions.

Figure 3: LC-MS chromatograms for both analytes and internal standard, after analysis of aqueous reference solution containing 9-cRA, 4-oxo-9-cRA and 13-cRA-d5.
Selection of internal standard
In LC-MS/MS analysis, selection of internal standard with similar chromatographic and mass spectrometric behavior to that of analyte is of utmost priority. The best internal standard is a stable-isotope form of the analyte which proves to be helpful when significant matrix effect is possible. Deuterated compound of 9-cRA and 4-oxo-9-cRA were not used as internal standards, due to their limited availability and high cost. Hence, isotretinoin-d5 (13-cRA-d5) was used as a common internal standard for both analyte.
Plasma extraction efficiency
Analytical methodology was optimized for extraction technique to attain consistent and high recovery with minimal matrix effect. Initially, the extraction of 9-cRA and 4-oxo-9-cRA was carried out via protein precipitation (with acetonitrile and methanol), but high back pressure with frequent clogging of the column was noted. Liquid-liquid extraction (LLE) technique was evaluated to isolate the drugs from human plasma using ethyl acetate, diethyl ether, n-hexane, dichloromethane, methyl tert-butyl ether and IPA (alone and in combinations) as extracting solvents. However, the recovery was inconsistent with significant ion suppression (>40%) in majority of extracting solvent mixtures used for LLE (Table 2). Finally, the combination of ethyl acetate-n-hexane-IPA (30:65:5, v/v/v) was found to be optimum as an extracting solvent mixture for high recovery with minimal ion suppression.
| Extracting solvent | (%) Process efficiency | (%) ion-suppression | ||
|---|---|---|---|---|
| 9-cRA | 4-oxo-9-cRA | 9-cRA | 4-oxo-9-cRA | |
| Ethyl acetate | 55.4 | 28.5 | 28.2 | 42.5 |
| Methyl tert-butyl ether | 42.7 | 22.5 | 42.8 | 37.8 |
| Diethyl ether | 25.7 | 14.5 | 25.8 | 30.5 |
| Diethyl ether:ethyl acetate (50:50, v/v) | 52.4 | 46.5 | 32.2 | 35.6 |
| Dichloromethane:n-hexane (50:50, v/v) | 45.5 | 25.8 | 28.6 | 38.5 |
| Dichloromethane:n-hexane:IPA (75:20:5, v/v) | 62.7 | 35.8 | 14.5 | 51.4 |
| Ethyl acetate:n-hexane (50:50, v/v) | 65.2 | 42.4 | 21.0 | 33.5 |
| Ethyl acetate:n-hexane:IPA (30:65:5, v/v/v) | 95.0 | 95.5 | 0.4 | 1.9 |
Table 2: Effect of extracting solvent on process efficiency and ion suppression.
During extraction optimization, extraction of 9-cRA and 4-oxo- 9-cRA from human plasma was carried out using LLE technique with acidic, neutral and alkaline medium. But in presence of acid and alkaline media, the hydrolysis of retinoyl-β-glucuronides and endogenous human blood compounds are induces which lead the higher RA concentrations [27]. In addition, poor extraction recovery was observed for both anlyte in acidic and alkaline pH. Extraction efficiency was improved in neutral pH and 300 μL of ammonium acetate buffer (pH 6.2) was selected as a pre-treatment solution. Inconsistency in peak area response of RAs and 13-cRA-d5 was observed during analysis of extracted samples. This could be due to low solubility of RAs and 13-cRA-d5 in the mobile phase that was finalized during chromatographic optimization. Low solubility of RAs and 13-cRA-d5 could be due to the high hydrophobic nature of these compound, which led to suppressed RAs and 13-cRA-d5 peak area response in the extracted samples. Therefore, the reconstitution solution composition was further optimized and it was observed that reconstitution solution, containing 5 mM ammonium acetate buffer (pH 9.0)-methanolacetonitrile (30:30:40, v/v/v) was suitable for solubility of RAs and 13-cRA-d5 and gave consistent 13-cRA-d5 peak area through out the analytical batch of larger sample size. Alkaline pH also improve the auto sampler stability (at 5°C) of RA.
Selectivity and signal-to-noise (S/N) ratio
There was no significant interference observed at the RT of 9-cRA, 4-oxo-9-cRA and 13-cRA-d5 in screened plasma lots. The mean interference observed at the retention time of the analyte between 10 different lots of human plasma including hemolyzed and lipemic plasma were calculated as 6.8%, 5.3% and 0.1% for 9-cRA, 4-oxo-9-cRA and 13-cRA-d5, respectively. Six replicates of LOQ samples were prepared from the cleanest blank samples and analyzed samples were deemed acceptable with %CV <5 for both analytes and internal standatds. The typical chromatograms of blank sample, blank processed with internal standard, LOQ, ULOQ and incurred sample in human plasma are shown in Figure 4. We observed that S/N ratio was >25 during method validation and clinical sample analysis, which was within acceptable limit as per the USFDA/EMEA guidelines.

Figure 4: Representative chromatograms of (A) double blank, (B) single blank processed with internal standard, (C) LOQ, (D) ULOQ and (E) incurred sample (Cmax) respectively (after 3.0 h of oral administration).
Linearity, precision and accuracy
The limit of quantitation in plasma was 2.85 ng/mL and 2.33 ng/ mL for 9-cRA and 4-oxo-9-cRA, respectively. The calibration curve was linear from 2.85-450.51 ng/mL and 2.33-180.57 ng/mL for 9-cRA and 4-oxo-9-cRA, respectively. Calibration curve was constructed using peak area ratio of analyte to internal standard and by applying linear, weighted least squares regression analysis with weighting factor of 1/ (concentration)2. The ‘r’ was greater than 0.99 for both analyte during the course of precision and accuracy batches. The correlation coefficient (r), slope and intercept value observed in three precision and accuracy batches are summarized in Table 3a. The intra-day precision and interday precision (%CV) ranged from 2.0-6.0 for 9-cRA and 5.7-13.8 for 4-oxo-9-cRA, respectively. The intra-day precision and accuracy at LOQQC level were 5.9% and 99.2%, respectively, for 9-cRA and 13.8% and 101.1%, respectively, for 4-oxo-9-cRA. The inter-day precision and accuracy at LOQQC level were 6.0% and 98.4%, respectively, for 9-cRA and 11.3% and 100.4%, respectively, for 4-oxo-9-cRA. The results of three precision and accuracy batches are summarized in Table 3b.
| Precision and accuracy batch No. | 9-cRA | 4-oxo-9-cRA | ||||
|---|---|---|---|---|---|---|
| Correlation coefficient (r) | Slope(m) | Intercept (c) | Correlation coefficient (r) | Slope (m) | Intercept(c) | |
| 1 | 0.9990 | 0.00574 | 0.00213 | 0.9972 | 0.00473 | 0.00168 |
| 2 | 0.9993 | 0.00571 | 0.00220 | 0.9955 | 0.00476 | 0.00206 |
| 3 | 0.9989 | 0.00633 | 0.00206 | 0.9984 | 0.00571 | 0.00218 |
| Mean | 0.99907 | 0.00593 | 0.00213 | 0.99703 | 0.00507 | 0.00197 |
| %CV | 0.02 | 5.90 | 3.29 | 0.15 | 11.00 | 13.23 |
Table 3a: Linearity data.
| Analyte name | QC sample | Nominal conc. (ng/mL) | Intra-run (n=6) | Inter-run (n=18) | ||||
|---|---|---|---|---|---|---|---|---|
| Mean observed conc.(ng/mL) | % CV | % Accuracy | Mean observed conc.(ng/mL) | % CV | % Accuracy | |||
| 9-cRA | LOQQC | 2.87 | 2.85 | 5.9 | 99.2 | 2.82 | 6.0 | 98.4 |
| LQC | 8.14 | 8.03 | 4.1 | 98.6 | 7.91 | 2.0 | 97.2 | |
| MQC | 189.29 | 188.09 | 2.9 | 99.4 | 187.43 | 3.8 | 99.0 | |
| HQC | 378.57 | 370.23 | 2.3 | 97.8 | 372.64 | 2.3 | 98.4 | |
| 4-oxo-9-cRA | LOQQC | 2.34 | 2.37 | 13.8 | 101.1 | 2.35 | 11.3 | 100.4 |
| LQC | 6.68 | 6.89 | 5.9 | 103.1 | 7.07 | 8.4 | 105.9 | |
| MQC | 75.94 | 73.85 | 5.7 | 97.3 | 75.87 | 6.0 | 99.9 | |
| HQC | 151.87 | 155.02 | 7.7 | 102.1 | 153.56 | 7.1 | 101.1 | |
Table 3b: Intra-and inter-day precision and accuracy data.
Process efficiency
The PE of 9-cRA, 4-oxo-9-cRA and 13-cRA-d5 were consistent across the QC levels. The mean PE of 9-cRA, 4-oxo-9-cRA and 13-cRA-d5 by the developed method were 96.8%, 95.1% and 91.6% respectively. The %CV of mean PE across the low, middle and high QC levels was <3 for 9-cRA and <4 for 4-oxo-9-cRA. The results of PE are presented in Table 4.
| Analyte Name | QC sample | Aa (%CV)b | Bc(%CV)b | Process efficiency(%PE)d |
|---|---|---|---|---|
| 9-cRA | LQC | 39797(3.3) | 40634(1.3) | 97.9 |
| MQC | 890967(4.2) | 943410(0.4) | 94.4 | |
| HQC | 1816689(1.3) | 1849191(0.6) | 98.2 | |
| 4-oxo-9-cRA | LQC | 38972(3.0) | 39546(4.9) | 98.5 |
| MQC | 387387(7.7) | 418743(0.9) | 92.5 | |
| HQC | 896161(1.3) | 922983(8.8) | 94.2 |
aMean area response of six replicate samples prepared by spiking before extraction (n=6)
bCoefficient of variation
cMean area response of six replicate samples prepared in reconstitution solution (n=6)
dA/B×100
Table 4: Process efficiency.
Matrix effect
Absolute matrix effect (AME) has a significant role in electro spray ionization mass spectrometry, which influence the ionization of analyte by ion-suppression or enhancement. The %CV of AME at each QC level were in the range of 0.9-1.6 and 0.7-1.9 for 9-cRA and 4-oxo- 9-cRA, respectively and between three QC levels it was 2.6 and 2.9 for 9-cRA and 4-oxo-9-cRA, respectively. As a result, the matrix effect from plasma was negligible for both analyte, followed by the extraction procedure of the method. The results of AME is presented in Table 5. The relative matrix effect (RME) was deemed acceptable, as presented in Table 6.
| Plasma lot | Absolute matrix effect | |||||
|---|---|---|---|---|---|---|
| 9-cRA | 4-oxo-9-cRA | |||||
| LQC | MQC | HQC | LQC | MQC | HQC | |
| Lot- 1 | 1.01 | 0.97 | 0.99 | 0.97 | 0.96 | 0.98 |
| Lot-2 | 1.00 | 0.98 | 1.02 | 0.97 | 0.97 | 1.03 |
| Lot-3 | 0.98 | 0.96 | 1.03 | 0.96 | 0.96 | 1.02 |
| Lot-4 | 1.00 | 0.96 | 1.02 | 0.98 | 0.96 | 1.03 |
| Lot-5a | 1.00 | 0.98 | 1.02 | 0.97 | 0.96 | 1.01 |
| Lot-6b | 1.02 | 0.97 | 1.04 | 0.97 | 0.95 | 1.00 |
| Mean | 1.00 | 0.97 | 1.02 | 0.97 | 0.96 | 1.01 |
| % CV | 1.3 | 0.9 | 1.6 | 0.7 | 0.7 | 1.9 |
aHemolyzed plasma lot;bLipemic plasma lot
Table 5: Absolute matrix effect.
| Plasma lot | Observed conc. (ng/mL) | |||
|---|---|---|---|---|
| 9-cRA | 4-oxo-9-cRA | |||
| LOQQC | HQC | LOQQC | HQC | |
| Lot- 1 | 2.68 | 361.43 | 2.30 | 151.78 |
| Lot- 1a | 2.79 | 375.32 | 2.27 | 153.37 |
| Lot- 2 | 2.56 | 390.77 | 2.42 | 144.64 |
| Lot- 2a | 2.67 | 358.56 | 2.40 | 142.91 |
| Lot- 3 | 2.86 | 352.71 | 2.25 | 145.48 |
| Lot- 3a | 2.58 | 357.69 | 2.38 | 146.30 |
| Lot- 4 | 2.79 | 371.07 | 2.30 | 152.53 |
| Lot- 4a | 2.65 | 385.25 | 2.06 | 144.35 |
| Lot- 5 | 2.63 | 356.89 | 2.36 | 139.95 |
| Lot- 5a | 2.93 | 358.12 | 2.52 | 146.95 |
| Lot- 6 | 2.71 | 356.56 | 2.11 | 141.89 |
| Lot- 6a | 2.86 | 360.02 | 2.32 | 145.13 |
| Mean | 2.73 | 365.37 | 2.31 | 146.27 |
| %CV | 4.4 | 3.4 | 5.5 | 2.9 |
| Nominal conc.(ng/mL) | 2.87 | 378.57 | 2.34 | 151.87 |
| %Nominal | 95.0 | 96.5 | 98.6 | 96.3 |
aDuplicate; Lot 5 is haemolyzed plasma;Lot 6 islipemic plasma
Table 6: Relative matrix effect.
Stability assessment
Stock solution stability of 9-cRA, 4-oxo-9-cRA and 13-cRA-d5 were established for 22 days at specified condition and % stability were 96.1%, 100.7% and 105.8%, respectively. 9-cRA and 4-oxo-9-cRA were proved to be stable in plasma for three freeze-thaw cycles. Bench top stability of both analyte were established for ~6.35 h in human plasma in ice cold water bath and under red light conditions. Auto sampler stability was assessed for ~128 h and long term stability was established at -50°C for 183 days. The observed mean concentration of both analyte was found to be within ± 15% of their respective nominal concentration and % CV was less than 15 at LQC and HQC levels (Table 7). 9-cRA and 4-oxo-9-cRA were found to be stable in human K3EDTA whole blood for ~2.45 h (Table 8).
| Storage conditions | 9-cRA | 4-oxo-9-cRA | ||||||
|---|---|---|---|---|---|---|---|---|
| Spikedconc. (ng/mL) | Mean observed conc. (ng/mL) | % CV | % change | Spiked conc. (ng/mL) | Mean observed conc. (ng/mL) | % CV | % change | |
| Bench top stability(~ 6.5 h, in ice cold water bath) | 8.14 | 8.16 | 4.7 | 0.25 | 6.68 | 6.71 | 5.5 | 0.45 |
| 378.57 | 377.45 | 1.4 | -0.30 | 151.87 | 150.23 | 5.6 | -1.08 | |
| Freeze-thaw stability(three cycles) | 8.14 | 8.21 | 3.5 | 0.86 | 6.68 | 6.76 | 7.6 | 1.20 |
| 378.57 | 390.58 | 3.3 | 3.17 | 151.87 | 153.21 | 3.9 | 0.88 | |
| Auto sampler stability(~ 128 h, at 5°C) | 8.14 | 8.30 | 2.2 | 1.97 | 6.68 | 6.76 | 4.3 | 1.20 |
| 378.57 | 378.23 | 2.4 | -0.09 | 151.87 | 149.23 | 2.9 | -1.74 | |
| Long term stability(128 days, below -50°C) | 8.14 | 8.40 | 0.9 | 3.19 | 6.68 | 6.81 | 3.2 | 1.95 |
| 378.57 | 392.57 | 2.2 | 3.70 | 151.87 | 152.30 | 3.5 | 0.28 | |
Table 7: Stability data (n=4).
| Analyte name | QC sample | Mean peak area ratio | (%) Stability | |
|---|---|---|---|---|
| Stability sample (%CV) | Comparison sample (%CV) | |||
| 9-cRA | LQC | 0.0458 (2.36) | 0.0470 (5.23) | 97.5 |
| HQC | 2.2126 (0.81) | 2.2280 (1.92) | 99.3 | |
| 4-oxo-9-cRA | LQC | 0.05078 (4.81) | 0.04918 (5.83) | 103.3 |
| HQC | 1.28350 (2.32) | 1.21385 (2.76) | 105.7 | |
Table 8: Whole blood stability (n=4).
Re-injection reproducibility and dilution integrity
Re-injection reproducibility exercise was performed to check whether the instrument performance remains unchanged after hardware deactivation because of any instrument failure during real clinical sample analysis. Percent difference for all re-injected QC samples was ≤ 7.29 for both analyte and deemed acceptable. The result of dilution integrity was deemed acceptable for 2 times and 4 times dilutions.
Carry over test evaluation
The MRM chromatograms of double blank (free from 9-cRA, 4-oxo-9-cRA and 13-cRA-d5) analyzed by following the ULOQ samples showed that there was no carry over.
Application to pharmacokinetic assessment
Following analysis, pharmacokinetic parameters like the maximum plasma concentration (Cmax), time to reach Cmax (Tmax), the half life (t1/2), the area under the plasma concentration-time curve (AUC0→t and AUC0→∞) were calculated by non-compartmental analysis using WinNonlin Professional software (Version 5.0, Pharsight Corp., Mountain View, CA, USA). The pharmacokinetic parameters are summarized in Table 9. The linear plot of mean plasma concentration (ng/mL) versus time (hr) for 9-cRA and 4-oxo-9-cRA are shown in Figure 5 and Figure 6, respectively.
| Analyte name | Formulation | PK Parameters | ||||
|---|---|---|---|---|---|---|
| Tmax (h) | Cmax (ng/mL) | AUC0→t (h.ng/mL) | AUC0→∞ (h.ng/mL) | t1/2 (h) | ||
| 9-cRA | R1 | 2.81 ± 1.25 | 210.20 ± 172.59 | 456.33 ± 281.43 | 522.88 ± 311.64 | 3.89±3.13 |
| R2 | 3.14 ± 1.26 | 203.02 ± 99.06 | 471.71 ± 192.27 | 494.98 ± 201.35 | 3.64±1.94 | |
| T | 3.04 ± 1.08 | 205.42 ± 121.70 | 451.85 ± 246.12 | 498.98 ± 239.16 | 3.35±3.07 | |
| 4-oxo-9-cRA | R1 | 2.96 ± 1.22 | 24.29 ± 22.67 | 38.55 ± 34.68 | 54.56 ± 29.94 | 0.59±0.31 |
| R2 | 3.24 ± 1.24 | 25.48 ± 17.60 | 40.56 ± 20.94 | 54.55 ± 23.48 | 0.61±0.27 | |
| T | 3.08 ± 1.10 | 29.01 ± 24.21 | 47.00 ± 22.54 | 77.26 ± 32.52 | 0.49±0.19 | |
R1 and R2 are the reference product; T is test product
Table 9: Pharmacokinetic parameters.

Figure 5: Plasma concentration profile. Linear plot of mean plasma concentration (ng/mL) versus time (hr) of 9-cRA; R1, R2: Reference drug; T: Test drug.

Figure 6: Plasma concentration profile. Linear plot of mean plasma concentration (ng/mL) versus time (hr) of 4-oxo-9-cRA; R1, R2: Reference drug; T: Test drug.
Conclusion
In summary, a rapid, selective, specific, reproducible and highthroughput LC-MS/MS method was developed and validated to estimate 9-cRA and 4-oxo-9-cRA in human plasma using 13-cRA-d5 as an internal standard. The proposed method showed good performance with respect to all the validation parameters tested, demonstrated optimized working conditions for both 9-cRA and 4-oxo-9-cRA human plasma with minimal inter-conversion and was successfully employed for a bioequivalence study of alitretinoin after oral administration of 30 mg capsule. It is possible with the described method for estimation of 9-cRA, 13-cRA and aTRA and their 4-oxo-metabolites simultaneously in human plasma.
Acknowledgements
All the authors wish to acknowledge the support and facilities received from Sun Pharmaceutical Industries Ltd., Gurgaon, India for carrying out this work.
References
- Packer L (1990) Retinoids, Part A: Molecular and Metabolic Aspects. Methods Enzymology. p: 189.
- Packer L (1990) Retinoids, Part B: Cell Differentiation and Clinical Aspects. Methods Enzymology. p: 190.
- Goss GD, McBurney MW (1992) Physiological and clinical aspects of vitamin A and its metabolites. Crit Rev Clin Lab Sci 29: 185-215.
- Ruzicka T, Lynde CW, Jemec GB, Diepgen T, Berth-Jones J, et al. (2008) Efficacy and safety of oral alitretinoin (9-cis retinoic acid) in patients with severe chronic hand eczema refractory to topical corticosteroids: results of a randomized, double-blind, placebo-controlled, multicentre trial. British Journal of Dermatology 158: 808-817.
- Petkovich M, Brand NJ, Krust A, Chambon P (1987) A human retinoic acid receptor which belongs to the family of nuclear receptors. Nature 330: 444-450.
- Giguere V, Ong ES, Segui P, Evans RM (1987) Identification of a receptor for the morphogen retinoic acid. Nature 330: 624-629.
- Mangelsdorf DJ, Ong ES, Dyck JA, Evans RM (1990) Nuclear receptor that identifies a novel retinoic acid response pathway. Nature 345: 224-229.
- Mangelsdorf DJ, Umesono K, Kliewer SA, Borgmeyer U, Ong ES, et al. (1991) A direct repeat in the cellular retinol-binding protein type II gene confers differential regulation by RXR and RAR. Cell 66: 555-561.
- Mangelsdorf DJ, Borgmeyer U, Heyman RA, Zhou JY, Ong ES, et al. (1992) Characterization of three RXR genes that mediate the action of 9-cis retinoic acid. Genes Dev 6: 329-344.
- Chen ZX, Xue YQ, Zhang R, Tao RF, Xia XM, et al. (1991) A clinical and experimental study on all-trans retinoic acid-treated acute promyelocytic leukemia patients. Blood 78: 1413-1419.
- Warrell RP Jr, Frankel SR, Miller WH Jr, Scheinberg DA, Itri LM, et al. (1991) Differentiation therapy of acute promyelocytic leukemia with tretinoin (all-trans-retinoic acid). N Engl J Med 324: 1385-1393.
- Kizaki M, Ikeda Y, Tanosaki R, Nakajima H, Morikawa M, et al. (1993) Effects of novel retinoic acid compound, 9-cis-retinoic acid, on proliferation, differentiation, and expression of retinoic acid receptor-alpha and retinoid X receptor-alpha RNA by HL-60 cells. Blood 82: 3592-3599.
- Adamson PC, Murphy RF, Godwin KA, Ulm EH, Balis FM (1995) Pharmacokinetics of 9-cis-retinoic acid in the rhesus monkey. Cancer Res 55: 482-485.
- Chomienne C, Ballerini P, Balitrand N, Daniel MT, Fenaux P, et al. (1990) All-trans retinoic acid in acute promyelocytic leukemias. II. In vitro studies: structure-function relationship. Blood 76: 1710-1717.
- Napoli JL (1986) Quantification of physiological levels of retinoic acid. Methods Enzymol 123: 112-124.
- Lanvers C, Hempel G, Blaschke G, Boos J (1996) Simultaneous determination of all-trans-, 13-cis- and 9-cis-retinoic acid, their 4-oxo metabolites and all-trans-retinol in human plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl 685: 233-240.
- Disdier B, Bun H, Catalin J, Durand A (1996) Simultaneous determination of all-trans-, 13-cis-, 9-cis-retinoic acid and their 4-oxo-metabolites in plasma by high-performance liquid chromatography. J Chromatogr B Biomed Appl 683: 143-154.
- Miyagi M, Yokoyama H, Shiraishi H, Matsumoto M, Ishii H (2001) Simultaneous quantification of retinol, retinal, and retinoic acid isomers by high-performance liquid chromatography with a simple gradiation. J Chromatogr B Biomed Sci Appl 757: 365-368.
- Rühl R, Schweigert FJ (2003) Automated solid-phase extraction and liquid chromatographic method for retinoid determination in biological samples. J Chromatogr B Analyt Technol Biomed Life Sci 798: 309-316.
- Dzerk AM, Carlson A, Loewen GR, Shirley MA, Lee JW (1998) A HPLC method for the determination of 9-cis retinoic acid (ALRT1057) and its 4-oxo metabolite in human plasma. J Pharm Biomed Anal 16: 1013-1019.
- Schmidt CK, Brouwer A, Nau H (2003) Chromatographic analysis of endogenous retinoids in tissues and serum. Anal Biochem 315: 36-48.
- Marchetti MN, Bun H, Geiger JM, Durand A (1994) Determination of a New Retinoid: 9-CIS Retinoic Acid in Plasma by HPLC. Analytical Letters 27: 1847-1862.
- Arnold SL, Amory JK, Walsh TJ, Isoherranen N (2012) A sensitive and specific method for measurement of multiple retinoids in human serum with UHPLC-MS/MS. J Lipid Res 53: 587-598.
- Wang Y, Chang WY, Prins GS, van Breemen RB (2001) Simultaneous determination of all-trans, 9-cis, 13-cis retinoic acid and retinol in rat prostate using liquid chromatography-mass spectrometry. J Mass Spectrom 36: 882-888.
- Wyss R (1990) Chromatography of retinoids. J Chromatogr 531: 481-508.
- USFDA (2001) Guidance for Industry: Bioanalytical Method Validation.
- European Medical Agency (2011) Guideline on bioanalytical method validation.
Relevant Topics
Recommended Journals
Article Tools
Article Usage
- Total views: 15157
- [From(publication date):
specialissue-2015 - Aug 29, 2025] - Breakdown by view type
- HTML page views : 13903
- PDF downloads : 1254