Research Article Open Access
Interplay between Peptide Length, Ionic Strength and pH in Electrophoretic eparations of Polyglutamate
Neeraja Venkateswaran1, Joshua M Kogot2 and Sumita Pennathur1*1Department of Biomolecular Science and Engineering, University of California, Santa Barbara, CA, USA
2Naval Surface Warfare Center, Panama City Division, Science and Technology Department, Panama City FL, USA
- *Corresponding Author:
- Sumita Pennathur
Department of Biomolecular Science and Engineering
University of California, Santa Barbara, CA, USA
Tel: 805-893-5510
E-mail: sumita@engr.ucsb.edu
Received date: October 11, 2015; Accepted date: November 03, 2015; Published date: November 10, 2015
Citation: Venkateswaran N, Kogot JM, Pennathur S (2015) Interplay between Peptide Length, Ionic Strength and pH in Electrophoretic Separations of Polyglutamate. J Anal Bioanal Tech S13:004. doi:10.4172/2155-9872.S13-004
Copyright: © 2015 Venkateswaran N, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Analytical & Bioanalytical Techniques
Abstract
A fundamental study of peptide transport behavior provides information about length dependent transport and sequence structure relationships. In these highly accurate investigations of peptide electrokinetic properties and peptide conformations using a Micro-Electro-Mechanical Systems (MEMS)-fabricated capillary zone electrophoresis (CZE) platform, we demonstrate accurate and repeatable separations of polyglutamate peptides of two different lengths (10 and 20 amino acids). We investigate this separation phenomenon as a function of electrolyte concentration and ionic composition, counter-ion radius, ionic strength and pH. We report a length-dependent counter-ion species selectivity and enhanced separation at pH much higher than the pKa of the peptides that can aid in understanding the relationship between the function of biological peptides and their microenvironments. The ability of our platform to optimize separation resolution between very similar peptides is promising for distinguishing molecular characteristics of peptides and can contribute to resolving current challenges in peptidomics.
Keywords
Capillary electrophoresis; Microfluidics; Peptides; Ionic strength; Persistence length; Electrostatics; Polyglutamate; Species selectivity
Introduction
Capillary electrophoresis (CE) techniques have been fundamental in advancing technology for the separation and identification of peptide and protein mixtures [1,2]. Today, they have become a staple in conjunction with mass spectrometry systems to resolve, identify and analyze protein and peptide characteristics with sensitivity and accuracy. Recent advances have miniaturized traditional CE instrumentation into microfluidic systems that can provide fast, highresolution separations with low sample volumes and better flow control [3]. However, the performance of traditional CE and microfluidic CE has rarely been compared. Furthermore, CE techniques such as on-chip gel electrophoresis, isotachophoresis [4], capillary isoelectric focusing [5] and capillary zone electrophoresis (CZE) have been applied from simple biomolecule separations to confirmatory diagnostics [6] and yet, rarely used for studying fundamental properties of peptides and proteins. In this way, CE has been underutilized as a stand-alone technique to probe structural and functional attributes of peptides.
Furthermore, the study of peptides, especially peptidomics has been on the rise since the early 2000’s and has become a prominent field in investigating the role of peptides in biochemical pathways. Initially an offshoot of proteomics to study proteolytic peptide fragments, peptidomics now spans the study of small peptides across many disciplines including [7,8], but not limited to neurological disorders [9], drug targeting vehicles [10], foodomics [11], and antimicrobial agents [12]. Critical challenges in peptidomic studies involve the identification of endogenous peptides [13] and tracing posttranslational modifications (PTMs) of proteins following proteolytic digestion. To date, a multitude of methods used for proteomics have been applied towards peptidomics, but peptidomic specific tools have also been invented and explored to target peptides [14].
In peptidomics, separation techniques such as 2D gel electrophoresis (2-DGE), high-performance liquid chromatography (HPLC), and capillary electrophoresis methods are combined with mass spectrometry (MS) techniques such as MALDI-TOF [2,11] and ESI [8] in separating and identifying peptides. Early separation techniques (2- DGE) are excellent in separating intact high molecular weight proteins with and without PTMs, but suffer in resolution when separating small proteins and peptide fragments [15]. To overcome this problem and to improve ease of integration with MS techniques, peptidomic technologies have moved to CE-MS systems to provide free solution separations of small peptides. However, the CE separation in CE-MS systems has been primarily used for obtaining peptide separation and not for investigating the electrostatic and conformational properties of peptides (which govern electrokinetic transport). In fact, a fundamental study of peptide transport behavior in chip-based CE could provide information regarding sequence structure relationships, length dependent transport and electrostatic screening.
We demonstrate a Micro-Electro-Mechanical Systems (MEMS)- fabricated CZE platform that can be used to measure electrokinetic properties to probe conformation and electrostatic changes with high accuracy in short, charged peptides. Our low-cost MEMS platform has advantages of small sample volumes and short analysis times. Furthermore, we can efficiently separate molecules within 2.5 cm compared to the traditional 30 cm capillaries used in CZE, with analysis times as short as 20 s (as opposed to ~20 minutes), limiting charge buildup and surface fouling. The microfluidic platform allows better control of applied electric fields and injection times while providing valuable information based on peptide-wall interactions, the electric double layer (EDL) surface phenomena and hydrodynamic interactions. To our knowledge, we are the first to perform a systematic study of short, charged, homopolymeric-peptides in a microfluidic capillary electrophoresis (MCE) platform to understand the conformational and electrostatic properties of peptides. We establish the repeatability and the efficacy of our platform by comparing it to traditional CE instruments and investigate the electrokinetic behavior of polyglutamate under different salt, pH and electrolyte composition. We use polyglutamate as a model system to begin our investigation into relating electrokinetic behavior with nanoscale phenomena. We observe a length dependent separation for polyglutamate with 10 and 20 residues (E10 and E20) that can be realized by differences in measured electrophoretic mobilities.
Additionally, changes in background electrolyte concentration cause mobility shifts induced by electrostatic screening of negatively charged residues. We observe preferential screening by larger counterions that enhance separation resolution between E10 and E20 and interplay between the ionic strength of the electrolyte and the pH in separation efficacy. As expected, separation resolution is lost at higher ionic strength as the negatively charged glutamate side-chains are screened by increasing concentrations of counter-ions, reducing their overall effective charge [16]. This behavior is parallel to that of double stranded DNA less than 400 base-pairs [17] though the upper limit of the number of residues required remains unexplored due to limitations in the synthesis of polyglutamate.
Materials and Methods
All chemicals were purchased from Sigma Aldrich. Sodium tetraborate (Na2B4O7) buffered solutions were made by combining equimolar concentrations of 100 mM boric acid (H3BO3) and 100 mM sodium tetraborate decahydrate (Na2B4O7.10H2O) to pH 9.21 measured with a pH meter (Oakton, Inc., WD-35634-30). No further titration with acid or base was required. Na2B4O7.10H2O and H3BO3 stock solutions were made using a 250 mL volumetric flask. Final concentrations varied from 10-50 mM Na2B4O7 supplemented with 0-90 mM added NaCl. Stock solutions of sodium phosphate (100 mM) were made using monobasic and dibasic sodium phosphate salts and titrated with 1M NaOH until the desired pH was reached. Stock solutions of NaCl and CsCl (100 mM) were made using a 250 mL volumetric flask and diluted to the appropriate concentrations for experiments. All solutions were filtered with 0.2 μm pore filters (Nalgene, Rochester, NY) prior to use.
Polyglutamate peptides
Polyglutamate sequences 10 and 20 residues in length were purchased from RSSynthesis in 1 mg aliquots. Peptides were labeled with FITC (fluorescein isothiocyanate) at the N-terminus. Stock samples were prepared by dissolving lyophilized peptides in deionized (DI) water at 1 mg/mL. Final samples were diluted down to 0.1 mg/mL (30 μM for E20, 60 μM for E10) in various concentrations of Na2B4O7 or at pH 9.21.
Capillary Electrophoresis (CE)
CE separations of E10 and E20 were performed on a P/ACE MDQ instrument (Beckman Coulter). Fused silica capillaries 30 cm in length with a 20 μm i.d. and 375 μm o.d. were used for all separations (Beckman Coulter). Effective length to detector was 20 cm. Note that the length could vary ± 1 cm as capillaries were trimmed down to appropriate length to fit in the capillary cartridge and required sanding of capillary ends to ensure symmetrical sample plugs. Samples were stored and separated at a temperature of 20°C. Applied electrophoresis voltage was 10 kV, corresponding to an electric field of 33.33 kV/m for a 30 cm capillary. Samples were detected using laser-induced fluorescence (LIF) with excitation at 488 nm and emission at 520 nm with a 3 mW argon ion laser. Each capillary was conditioned according to manufacturer’s instructions with methanol, water, 0.1M HCl, 0.1M NaOH and appropriate background electrolyte prior to running separation experiments. Fluorescein (10 μM) was added to all samples as an internal standard.
Microfluidic experimental setup
Microfluidic channels were custom designed and made using MEMS fabrication techniques on borosilicate wafers (Dolomite Inc.). Channel geometry consisted of a simple cross channel with four reservoirs, labeled North (N), South (S), East (E) and West (W) (Figure 1). Channels were 1 μm deep and 9 μm wide with 5 mm long N, S and W channels and a 30 mm long E separation channel. Electric fields were applied using platinum electrodes and a high voltage power supply (HVS448, Labsmith), using pre-programmed sequences for sample loading, gating and separation schemes (Figure 1). Samples were loaded from top to bottom (N-S reservoirs) and were gated from N-E. Following the injection of a small plug of sample into the east channel, separation voltages were applied to drive electrokinetic flow from W-E, enabling the separation of sample components in the east channel.

Figure 1: (Top) Experimental setup of MEMS CZE platform. (Bottom) Sample electropherogram of peptides E10 and E20 in Na2B4O7 with a 40.3 kV/m applied electric field, 25 mm downstream of junction.
Prior to separations, the channel was prepared by washing with water, 0.1M HCl, 0.1M NaOH, and appropriate background electrolyte. Separation E-field were set to 40.3 kV/m. Samples were imaged 25 mm downstream of the cross junction with an inverted epifluorescence microscope (Olympus IX70, Olympus Inc.) equipped with a 60X water immersion objective lens (1.0 Numerical Aperature, Olympus Inc.). A 473 nm laser attenuated to <10 mW was used as the excitation source along with a FITC fluorescence filter cube (467 nm to 498 nm excitation wavelength) and a dichroic mirror to detect the fluorescence emission of the sample plugs passing the detector.
Electrophoretic mobility measurements via electrokinetic transport behavior
Electrophoretic mobility of the E10 and E20 peptides were calculated using direct time-to-arrival measurements during electrokinetic injections as described by our group [18]. Briefly, the observed velocity of the samples was calculated from the raw data collected via fluorescence imaging and corresponds to the combination of the electro osmotic flow (EOF) and the electrophoretic velocity of the sample. EOF was calculated from current monitoring experiments previously described [19] and is subtracted from the observed velocity to determine the electrophoretic velocity of the sample. Finally, the electrophoretic mobility of the sample is determined by the ratio of the sample electrophorectic velocity to the electric field.
Results and Discussion
Figure 2 shows elution profiles of E10 and E20 in various concentrations of sodium tetraborate (Na2B4O7, pH 9.21) using both a traditional CE instrument (P/ACE MDQ, Beckman Coulter) and our custom microchip electrophoresis setup. The E10 and E20 polypeptides have isoelectric points of 3.29 and 3.05 respectively [20], and will remain negative throughout the course of our experiments. Note that these anionic peptides also reduce surface fouling of the negatively charged glass channel and capillary walls, allowing for repeatable and accurate data collection. Electrophoretic mobility measurements from our MCE setup (Figure 1) were compared to data collected from the P/ACE MDQ instrument to determine the efficacy of our platform in reproducibly measuring the mobility of peptides.
The data collected differed by less than 5% between CE and MCE and this small deviation can be attributed to slight variations in the running buffer composition [21]. Note that the electrophoretic mobility of E20 was greater than E10 at low electrolyte concentration. The monotonic increase in electrophoretic mobility of E10 and E20 (Figure 2) can in part be explained by the relationship between the thickness of the EDL and counter-ion concentration. As the surrounding electrolyte concentration increases, the debye screening length surrounding charged polyelectrolytes decreases lowering the effective charge on the peptide [22]. Increasing the background electrolyte concentration increases the availability of counterions in solution to screen the carboxylate side chains and therefore, results in an increase in the electrophoretic mobility of E10 and E20.

Figure 2: Measured electrophoretic mobilities of E10 and E20 in a 20 μm capillary (CE) and in a 1 μm microfluidic channel (1 μm) as a function of increasing Na2B4O7.
Furthermore, we observe that MCE results are more accurate and reproducible than CE experiments. The reproducibility of CE data suffers possibly due to the charge build up in the capillary walls over long analysis times and unequal buffer volumes in the reservoirs, which can in turn cause pressure driven flow based dispersion [23]. In addition, zeta potentials can vary at the capillary surface [24], which changes the electroosmotic flow over the course of a single experiment. This variation is noted by the high standard deviations in CE measurements and inability to distinguish differences in the electrophoretic mobility of E10 and E20 as a function of electrolyte concentration (Figure 2, CE). In contrast, consecutive MCE experiments can be performed quickly in succession (<30 seconds) preventing charge buildup on the channel walls and thus variations in both zeta potential and flow.
Electrolyte concentration
The electrophoretic mobility measurements obtained from our microfluidic setup clearly differentiate the electrokinetic behavior of the two peptides. The differences between the same peptide in a CE vs. an MCE setup are around the order of 2.5% in low electrolyte concentrations, decreasing to 0.2% at higher concentrations (Figure 2) (calculated by subtracting the two numbers and dividing by the largest). This separation phenomenon has not previously been observed for polyglutamate in free solution CZE, though the majority of electrokinetic studies in free solution are limited to longer lengths of polyglutamate [25,26]. Thus, MCE can provide insight into subtle conformational and electrostatic mechanisms that govern the differences observed in electrophoretic mobility. Importantly, MCE can be utilized in studying the relationship between peptide mobility, localized charge density, sequence-structure relationships and nearest neighbor effects as a function of increasing peptide length. Ultimately, insight into these phenomena will aid in discerning details of protein structure and function.
To understand what mechanisms govern the differences observed in mobility between the two peptides, we first note the charge to mass ratios of E10 and E20 differ by 0.1 m/z due to the additional negative charge contributed by FITC at the N-terminus. However, this does not account for the discrepancies seen in experimentally obtained electrophoretic mobilities (~2.5% in 20 mM Na+), suggesting differences in either conformation (which affects the frictional coefficient) and/ or the charge of the EDL. Furthermore, because at higher Na+, the difference in electrophoretic mobility reduces to 0.2% (Figure 2), both peptides likely obtain similar conformational and electrostatic characteristics at higher ionic strengths. Therefore, the data indicate a regime where the electrokinetic behavior is largely governed by either conformation and/or electrostatics, beyond which differences in electrokinetic mobility become negligible.
Experimentally, for E10 and E20, the measured electrophoretic mobilities varied more than calculated theoretical mobility approximations. Estimating radius of gyration (on the assumption that polyglutamates are a random coil in aqueous solution) and approximating a frictional coefficient based on a hard sphere of equivalent radii, the theoretical mobilities for E10 and E20 have no difference (-1.19 × 10-7 m2/Vs), not accounting for suppressed charge. The experimental mobility values for E10 and E20 are -4.80 × 10-8 m2/ Vs and -4.74 × 10-8 m2/Vs respectively in 20 mM Na+. The difference in experimental mobilities and the order of magnitude difference between theoretical and experimental values indicate that there are short range and long-range electrostatic forces that govern electrokinetic transport [27] that cannot be account for purely by theoretical mobility approximations. When examining modified models proposed by Offord et al. and Grossman et al. [28], we calculate mobilities that are the same order of magnitude as those experimentally derived. However, these models often take into account a parametric constant that is based on experimental conditions, specifically electrostatics, shape effects, pH and species selectivity. The following MCE experiments explore these phenomena individually to understand the contributions to the separation behavior observed.
Literature reports of the conformational properties of E10 and E20 state that polyglutamate is known to adopt an extended structure at basic pH due to the repulsion between neighboring glutamate residues (though it is reported that to adopt helical motifs at pH <4.5) [29,30]. To confirm the conformational properties with our experimental conditions, we measured the Circular Dichroism (CD) spectra of E10 and E20 at various electrolyte concentrations. Our experimental results are in accordance with previously reported literature and do not indicate any presence of helicity or other structural motifs at pH 9.21 as quantified by CDPro analysis [31]. Given that polyglutamate is strongly negatively charged at pH 9.21, the electrostatic screening of the residues most likely increases the flexibility of the backbone, which would increase the electrophoretic retardation force that would act upon it. Therefore, the combined effects of decreased electrostatic forces and increased retardation forces can result in a higher (more positive) electrophoretic mobility at higher electrolyte concentration.
Species selectivity
Variances in the EDL counterion composition surrounding the peptide are likely contributors to the differences in electrokinetic behavior between E10 and E20. The electrophoretic mobility of a molecule is governed by its charge-to-drag ratio and the effective charge on a molecule is dependent on the composition of the surrounding EDL. In Figure 3, we show elution profiles of E10 and E20 in 10 mM Na2B4O7 supplemented with either 10 mM NaCl (3A) or 10 mM CsCl (3B), two cations of varying ionic radius. As the peak shapes indicate, E10 and E20 resolved more distinctively in the buffer with added 10 mM CsCl compared to 10 mM NaCl, proving that electrostatics are very important in determining the electrokinetic behavior of these peptides. Note that the slight asymmetry seen in peak shapes for Figure 3B may be due to E10 and E20 co-entrapping Cs+ though this requires further investigation regarding the complexation of alkali metal ions with polyanionic peptides with respect to ionic radius.

Figure 3: Electropherograms of E10 and E20 separations in (A) 10 mM Na2B4O7 with 10 mM NaCl and (B) 10 mM Na2B4O7 with 10 mM CsCl. Detector placed 10 mm downstream of junction in a 1 μm microfluidic channel.
Species selectivity occurs in electrolytes that have different counterions that vary in size or valence [16], with polyions preferring small counterions for screening [16]. However, greater differences in electrophoretic mobility due to CsCl indicate that equilibrium may exist between Na+ and Cs+ ions in the EDL of E10 and E20. It is also theorized that larger counter-ions with the same valence do not screen charges as effectively due to steric hindrances and inefficient shortrange electrostatics [32]. E10 and E20 have lower electrophoretic mobilities and elute much later with 10 mM CsCl, suggesting the inability of Cs+ to screen the glutamate residues effectively. This trend is parallel to those seen with systematic studies of DNA with different sizes of counter-ions and their effect on the electrophoretic mobility [33].
To quantify the differences in behavior between the two electrolytes, we define a dimensionless parameter Q (equation 1), which represents the percent difference in electrophoretic velocities between the internal standard (IS) and the peptide of interest (i) in different electrolytes (EI and EII).
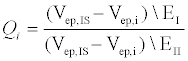
In the above equation, i represents E10 or E20, IS represents fluorescein, EI and EII indicate Na2B4O7 supplemented NaCl and CsCl respectively and Vep is the electrophoretic velocity of the species. If electrokinetic behavior of E10 and E20 are equally affected due to the addition of a larger counter ion such as Cs+, QE10 and QE20 would be expected to have the same value. However, this is not the case experimentally: QE10 has a value of 0.41 while QE10 has a value of 0.47. In the range 0<Q<1, a lower value of Q indicates that the EDL composition of E10 changes more than E20. Likely, the ineffective screening of charges on E10 is due to short range electrostatic forces that allow for more Cs+ ions to occupy the EDL or the propensity of E20 in attracting and accommodating a greater number of small counterions (Na+) next to the backbone. Either scenario would result in a greater negative change in the electrophoretic mobility of E10. Nevertheless, the manner in which larger counter-ions affect the electrophoretic mobility of E10 and E20 are an indication of length-dependent screening mechanisms that contribute to differences seen in electrophoretic mobility.
Electrolyte composition
In order to ensure that the differences in electrophoretic mobility are independent of the type of co-ion, we measured the electrokinetic mobility of E10 and E20 in 10 mM Na2B4O7 with varying amount of added NaCl to adjust the ionic strength (Figure 4). Additionally, Na2B4O7 is known to be insoluble at higher concentrations (above 100 mM), and most molecular dynamic simulations use monovalent salts to model peptide environments. However, since it is necessary to use a buffer to maintain the pH in electrophoretic experiments, using NaCl to increase the electrolyte concentration (with a nominal buffer concentration in the background) can help mimic simulation environments where the solvent is composed solely of salt ions [34]. Elution profiles were similar to those shown in Figure 2, with the electrophoretic mobility of E10 and E20 increasing with electrolyte concentration. However, the addition of NaCl to 10 mM Na2B4O7 decreases the electrophoretic mobility of E10 and E20 compared to mobilities measured in Na2B4O7 alone due to differences in the concentration and size of co-ions. In electrolytes with ~40 mM Na+ the EOF under an applied electric field of 40.3 kV/m for the electrolyte with supplemented NaCl is lower (2.47 mm/s) than for the electrolyte without NaCl (2.55 mm/s). Despite having the same concentration of counterions that drive fluid flow and screen the glutamate charges, E10 and E20 exhibit more negative electrophoretic mobilities in the electrolyte with NaCl.

Figure 4: Measured electrophoretic mobilities of E10 and E20 in a 1 μm microfluidic channel with 10 mM Na2B4O7 and increasing concentrations of NaCl.
Examining the co-ion composition in these two electrolytes reveals a 1:2 tetraborate to chloride ion ratio summing up to 30 mM co-ion concentration for the electrolyte with NaCl. With the 100% tetraborate ion composition, the co-ion concentration is only at 20 mM. Therefore, in the case of the NaCl enhanced electrolyte, there are a greater number of co-ions flowing the same direction as E10 and E20, opposing the electric field. This enhances the electrophoretic transport of E10 and E20, resulting in a more negative electrophoretic mobility as observed. Secondly, the presence of the borate ions in the hydration shells of the peptides may hinder the electrokinetic transport of E10 and E20 against the electric field due to the increased ionic radius of tetraborate. If so, the electrophoretic mobility with tetraborate would be greater in value, indicating slower transport behavior. However, the latter is unlikely as the mobility of the internal standard, fluorescein (whose hydration shell is significantly smaller) is equally reduced with added NaCl. Nonetheless, as we increase the concentration of the electrolyte, the increasing trend of electrophoretic mobility with either electrolyte remains valid as Cl- and B2O72- produce the same behavior. Therefore, although co-ion composition makes a difference in the absolute mobility measured, major trends remain the same.
An interesting trend seen in both data sets is that the electrophoretic mobility not only converged to the same values as the electrolyte concentration increases, but E10 and E20 also reversed their order of elution at the highest concentrations. This “reversal” in elution order may be due (1) electrostatic effects or (2) conformational differences. If dependent on electrostatics, the rate of charge neutralization on E10 may be faster than that of E20. If so, as electrolyte concentration increases, E10 would have a smaller charge to drag ratio, allowing it to elute first. If the “reversal” is dependent on conformational changes, E20 may be compacting at a faster rate than E10 as the longer length of E20 may provide more flexibility than E10. This would decrease the drag on E20, and would increase the charge to drag ratio compared to E10, causing E20 to elute second as electrolyte concentration increases. However, it is more likely that this “reversal” is also governed by differences in electrostatic screening that are length dependent as polyglutamate is expected to maintain a rod like shape in aqueous solutions.
Additionally, in an attempt to completely eliminate any effect the tetraborate ions may have in causing the separation, separations of E10 and E20 were performed at one concentration in pure 20 mM NaCl at pH 9.2 (titrated with NaOH). Elution profiles were similar to those seen with 10 mM Na2B4O7 (same cation concentration) as the electrolyte. However, due to the instability of the CE platform under non-buffered conditions and its propensity for acquiring pH gradients, these experiments were difficult to reproduce.
Ionic strength and pH
Furthermore, electrokinetic separations of E10 and E20 were performed in sodium phosphate buffer, whose ionic composition is well understood. Combined with the previous data, this gives a true picture of E10 and E20 behavior regardless of ionic composition and ionic strength. We performed separations of E10 and E20 at pH 7.5, pH 9.2 and pH 11.5 which corresponded to ionic strengths of 270 mM, 299 mM and 455 mM respectively (Figure 5). Sample plugs were injected downstream of the junction for the same elution time (but not detection distance) in order to minimize differences in peak shape due to diffusion. The lowest ionic strength of 270 mM (Figure 5A) gave the best resolution between peaks at all pHs. Resolution rapidly declines as a function of increasing ionic strength and decreasing pH though separations sustain good resolution at pH 11.5 regardless of peak broadening at all ionic strengths. Therefore, the elution profiles are indicative of a strong pH dependence on the transport behavior of E10 and E20, which is surprising given that the pH of the electrolytes are far from the pKa of the peptides (pKa~3.1). A moderate dependence with ionic strength is also seen though this trend breaks down with a rise in pH. The effect of pH and ionic strength of the electrolyte on the separation behavior suggests different protonation states for neighboring residues influenced by EDL composition and nearest neighbor effects [35]. This may induce subtle conformational differences between E10 and E20 that contribute significantly to the observed length dependent mobilities of polyglutamate.

Figure 5: Electropherograms of E10 and E20 separations at ionic strengths (A) 270 mM (B) 299 mM and (C) 455 mM at pH 11.5 (Top), pH 9.2 (middle) and pH 7.5 (bottom) in sodium phosphate buffer. Detection length varied to match elution times at each ionic strength.
Finally, we hypothesize that the different persistence lengths of the two peptides may play a role in their differing electrokinetic behavior. In literature, models of bent rod DNA have shown to have small but significant differences in electrophoretic mobility compared to rigid rod DNA models [36]. Likely, a similar phenomenon could be occurring with polyglutamate given the greater innate flexibility of peptides as compared with double stranded DNA. Persistence lengths of peptides vary widely due to their dependence on the peptide sequence, charge density and the presence of neighboring charge groups. At low [Na+], E10 may be within the limits of the persistence length due to the negatively charged side-chain repulsion and maintains a rod-like shape. This would allow it to have a decreased retardation force if E10 is capable of aligning to the direction of the flow.
Though it is probable that E20 remains in an extended conformation, its longer length may allow for more flexibility, which may result in an increased retardation force. However, as the electrolyte concentration increases, the charges on the E10 side-chains are screened due to counter-ion condensation, which may allow it to gain some flexibility, leading to the convergence of mobilities seen in Figure 2. The “reversal” of elution times between E10 and E20 at 110 mM Na+ can be hypothesized to occur for the following reasons. First, the carboxylates on the glutamate side chains on E10 may be neutralized faster than E20, resulting in E10 having an increased m/z ratio, increasing the flexibility beyond that of E20. Second, the increased flexibility of E10 may no longer allow it to align to the flow, causing a tumbling motion that increases the retardation force on the peptide while E20 remains aligned to the flow due to its greater concentrations of charges. Given the inherent difficulties with exploring flow-dependent alignment experimentally, such theories prompt future simulations of rod like polymers in electrokinetic transport systems.
Conclusion
Microfluidic capillary electrophoresis (MCE) has the potential to improve on the efficacy of traditional CE separations by providing repeatable, accurate and fast analysis of peptides. We demonstrate the ability of MCE to elucidate electrostatic phenomena that govern the length-dependent separation of E10 and E20, giving insight into the nanoscale physics of EDL and electrostatic screening. Sensitivity and resolution of separation can be optimized at pHs that are far removed from the pKa of peptides, for example, both at low ionic strengths and basic buffers for acidic peptides (E10 and E20). Furthermore, E10 and E20 demonstrate differences in species selectivity of counterions that are likely due to length-dependent polymer flexibility and end fraying effects, previously unexplored by any sort of separation-based tool. Thus, our work allows for the potential to delve deeper into the transport behavior of peptides while bringing the unexplored strengths of MCE into the forefront of peptidomic technologies.
Supporting Information
Supporting information Available: Quantitative comparison between P/ACE MDQ and MCE, EOF measurements in 1 μm channels, CD spectras of E10 and E20, separation of E10 and E20 in an unbuffered system and measured mobilities of fluorescein (internal standard). This material is available free of charge via the Internet at http://pubs.acs.org
Acknowledgements
This work was supported by the Institute for Collaborative Biotechnologies through grant W911NF-09-0001 and W911NF-12-1-0031 from the U.S. Army Research Office in addition to an ICB-CQL fellowship from the American Society for Engineering Education, and an Office of Naval Research In-house Laboratory Independent Research project. The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. The authors would like to thank R.V. Pappu and R.K. Das for stimulating discussions regarding peptide conformation.
References
- Kasicka V (2010) Recent advances in CE and CEC of peptides (2007-2009). Electrophoresis 31: 122-146.
- Stutz H (2005) Advances in the analysis of proteins and peptides by capillary electrophoresis with matrix-assisted laser desorption/ionization and electrospray-mass spectrometry detection. Electrophoresis 26: 1254-1290.
- Napoli M, Eijkel JC, Pennathur S (2010) Nanofluidic technology for biomolecule applications: a critical review. Lab Chip 10: 957-985.
- Miyazaki H, Katoh K (1976) Isotachophoretic analysis of peptides. J Chromatogr 119: 370-383.
- Herr AE, Molho JI, Drouvalakis KA, Mikkelsen JC, Utz PJ, et al. (2003) On-chip coupling of isoelectric focusing and free solution electrophoresis for multidimensional separations. Anal Chem 75: 1180-1187.
- Araz MK, Apori AA, Salisbury CM, Herr AE (2013) Microfluidic barcode assay for antibody-based confirmatory diagnostics. Lab Chip 13: 3910-3920.
- Menschaert G, Vandekerckhove TT, Baggerman G, Schoofs L, Luyten W, et al. (2010) Peptidomics coming of age: a review of contributions from a bioinformatics angle. J Proteome Res 9: 2051-2061.
- Herrero M, Ibañez E, Cifuentes A (2008) Capillary electrophoresis-electrospray-mass spectrometry in peptide analysis and peptidomics. Electrophoresis 29: 2148-2160.
- Westman-Brinkmalm A, Ruetschi U, Portelius E, Andreasson U, Brinkmalm G, et al. (2009) Proteomics/peptidomics tools to find CSF biomarkers for neurodegenerative diseases. Front Biosci (Landmark Ed) 14: 1793-1806.
- Arap W, Pasqualini R, Ruoslahti E (1998) Cancer treatment by targeted drug delivery to tumor vasculature in a mouse model. Science 279: 377-380.
- Simó C, Domínguez-Vega E, Marina ML, García MC, Dinelli G, et al. (2010) CE-TOF MS analysis of complex protein hydrolyzates from genetically modified soybeans--a tool for foodomics. Electrophoresis 31: 1175-1183.
- Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415: 389-395.
- Baggerman G, Verleyen P, Clynen E, Huybrechts J, De Loof A, et al. (2004) Peptidomics. J Chromatogr B Analyt Technol Biomed Life Sci 803: 3-16.
- Shen Y, Tolic N, Zhao R, Purvine SO, Schepmoes AA, et al. (2011) Effectiveness of CID, HCD, and ETD with FT MS/MS for Degradomic-Peptidomic Analysis: Comparison of Peptide Identification Methods. J Proteome Res 10: 3929-3943.
- Sewald N, Jakubke HD (2009) Peptide synthesis. (2nd edn) Peptides: Chemistry and Biology. pp: 175-315.
- Manning GS (1984) Limiting laws and counterion condensation in polyelectrolyte solutions. 8. Mixtures of counterions, species selectivity, and valence selectivity. J Phys Chem 88: 6654-6661.
- Stellwagen NC, Bossi A, Gelfi C, Righetti PG (2001) Do orientation effects contribute to the molecular weight dependence of the free solution mobility of DNA? Electrophoresis 22: 4311-4315.
- Pennathur S, Santiago JG (2005) Electrokinetic transport in nanochannels. 2. Experiments. Anal Chem 77: 6782-6789.
- Driehorst T, O’Neill P, Goodwin PM, Pennathur S, Fygenson DK (2011) Distinct Conformations of DNA-Stabilized Fluorescent Silver Nanoclusters Revealed by Electrophoretic Mobility and Diffusivity Measurements. Langmuir 27: 8923-8933.
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, et al. (2003) ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31: 3784-3788.
- VanOrman BB, Liversidge GG, McIntire GL, Olefirowicz TM, Ewing AG (1990) Effects of buffer composition on electroosmotic flow in capillary electrophoresis. J Microcolumn Sep 2: 176-80.
- Stigter D, Dill KA (1990) Charge effects on folded and unfolded proteins. Biochemistry 29: 1262-1271.
- Ghosal S (2006) Electrokinetic flow and dispersion in capillary electrophoresis. Annu Rev Fluid Mech 38: 309-338.
- Kirby BJ, Hasselbrink EF Jr (2004) Zeta potential of microfluidic substrates: 1. Theory, experimental techniques, and effects on separations. Electrophoresis 25: 187-202.
- Dolník V, Novotny MV (1993) Separation of amino acid homopolymers by capillary gel electrophoresis. Anal Chem 65: 563-567.
- Dolník V, Novotny M, Chmelík J (1993) Electromigration behavior of poly-(L-glutamate) conformers in concentrated polyacrylamide gels. Biopolymers 33: 1299-1306.
- Janini GM, Metral CJ, Issaq HJ, Muschik GM (1999) Peptide mobility and peptide mapping in capillary zone electrophoresis. Experimental determination and theoretical simulation. J Chromatogr A 848: 417-433.
- Adamson NJ, Reynolds EC (1997) Rules relating electrophoretic mobility, charge and molecular size of peptides and proteins. J Chromatogr B Biomed Sci Appl 699: 133-147.
- Giacometti G, Turolla A, Boni R (1968) Enthalpy of helix-coil transitions from heats of solution. I. Polyglutamates. Biopolymers 6: 441-448.
- Rai JH, Miller WG (1973) Electrostatic interactions in polyglutamic acid. Biopolymers 12: 845-856.
- Sreerama N, Woody RW (2000) Estimation of Protein Secondary Structure from Circular Dichroism Spectra: Comparison of CONTIN, SELCON, and CDSSTR Methods with an Expanded Reference Set. Anal Biochem 287: 252-260.
- Bohinc K, Kralj-Iglic V, Iglic A (2001) Thickness of electrical double layer. Effect of ion size. Electrochim Acta 46: 3033-3040.
- Ross PD, Scruggs RL (1964) Electrophoresis of DNA. III. The effect of several univalent electrolytes on the mobility of DNA. Biopolymers 2: 231-236.
- Tran HT, Mao A, Pappu RV (2008) Role of backbone-solvent interactions in determining conformational equilibria of intrinsically disordered proteins. J Am Chem Soc 130: 7380-7392.
- Schwartz H, Pritchett T (1994) Separation of Proteins and Peptides by Capillary Electrophoresis. Beckman Coulter.
- Allison S, Chen C, Stigter D (2001) The length dependence of translational diffusion, free solution electrophoretic mobility, and electrophoretic tether force of rigid rod-like model duplex DNA. Biophys J 81: 2558-2568.
Relevant Topics
Recommended Journals
Article Tools
Article Usage
- Total views: 12320
- [From(publication date):
specialissue-2015 - Aug 30, 2025] - Breakdown by view type
- HTML page views : 11338
- PDF downloads : 982