Research Article Open Access
Protein Gelation around Axons Inhibits Action Potential Propagation in Nerve Fibers
Wade N Dauberman, Samuel Breit and Shaohua Xu*Department of Biological Sciences, Florida Institute of Technology, 150 West University Blvd, Melbourne, FL 32901, USA
- *Corresponding Author:
- Shaohua Xu
Department of Biological Sciences
Florida Institute of Technology
150 West University Blvd, Melbourne
FL 32901
Tel: 321-674-8430
E-mail: shaohua@fit.edu
Received date: June 29, 2017; Accepted date: July 06, 2017; Published date: July 13, 2017
Citation: Dauberman WN, Breit S, Xu S (2017) Protein Gelation around Axons Inhibits Action Potential Propagation in Nerve Fibers. J Alzheimers Dis Parkinsonism 7:349. doi:10.4172/2161-0460.1000349
Copyright: © 2017 Dauberman WN, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Alzheimers Disease & Parkinsonism
Abstract
Recently, we reported that amyloid fibers can further aggregate and form gels. In this paper, we provide evidence that protein gels, when formed outside nerve fibers, can substantially reduce the compound action potential. Protein gelation also increases the viscosity of the media. The nerve fiber’s compound action potential was found to be inversely related to the concentration of glycerol applied extracellularly. Soluble oligomer aggregates and fibrils on the other hand had little effect on action potential. These results suggest that the formation of protein gels surrounding neuronal processes, as in the case of amyloid plaques of Alzheimer’s disease, may disrupt the propagation of action potential and then trigger a cascade of events leading to neuronal death. As illustrated in Darcy’s law, gels restrict fluid flow and then the circulation of ions and molecules, which might underlie the pathogenesis of Alzheimer’s disease.
Keywords
Self-assembly; Biogel; Transportation phenomena; Bulk flow; Synapse loss; Degenerating neurites
Introduction
Protein self-assembly and formation of amyloid fibers is an early event of numerous human diseases, including Alzheimer’s disease (AD) and Parkinson’s disease [1-3]. In AD, amyloid-beta (Aβ) peptides aggregate and form fibers which then either bundle or randomly tangle together and deposit extracellularly in the form of plaques [4,5]. These insoluble plaques bury inside a large number of degenerating neurites and are commonly associated with synapse loss and neuronal death [6]. How and whether the plaques or the aggregation intermediates are responsible for the cellular damage remain unclear.
Oligomers, or what we call colloidal spheres, are produced when proteins aggregate and form amyloid fibers. These fibers and colloids are proposed by different groups to be responsible for neuronal death. We found it problematic to name either the fibers or the colloids as the culprit. It is difficult to propose a plausible mechanism for how amyloid fibers damage cells, when fibers by other proteins such as collagen are everywhere in the body. Compared to the fibers, the colloids as the pathogen are broadly investigated over the last decade; but their cellular effect appears to be dependent on the experimental system used by different laboratories [7]. Also, some consider the colloids a “far cry” for being responsible for the massive neuronal loss found in AD [8].
In vitro, amyloid fibers have been found to be capable of further aggregating and forming gels [9,10]. Can amyloid plaques in AD be gels of porous matrix? TEM images of amyloid plaques either in AD tissue or isolated by density gradient centrifugation reveal these plaques are biogels with dense fiber network [3,5,10]. Gels are known to be capable of eliminating bulk flow and reduce diffusion. Darcy’s law has been developed for the analysis of fluid flow under pressure through a porous matrix including gels. A simplified version called Darcy’s flux states that:

where q is the flux of fluid per unit area through a porous matrix; κ is the Darcy’s permeability coefficient, ∇p is the hydrostatic pressure gradient vector across the matrix; and η is the viscosity of the cerebral spinal fluid inside the matrix. Thus, flux is strongly dependent on the Darcy’s permeability and the viscosity of the media. How does gelation affect κ and μ?
We hypothesize that gel formation around axons may inhibit the heart-pulse-driven fluid flow and then the circulation of nutrients and ions around the axon and affect the propagation of the action potential. In this paper, protein gels of fibrin were examined for their effect on the compound action potential (CAP) of Xenopus laevis sciatic nerves. The sciatic nerve is easy to isolate and shares common physiological features of neurons in the central nervous system [11]. The nerve fibers remain functional for 4 h in Ringer’s solution, which is long enough to test gelation effect on action potential. The limited lifetime of 4 h for an isolated nerve fiber prevents our use of amyloid fiber gels for the study of gels’ effect on CAP. It takes over three days for amyloid fibers to form gels and only 1 min for fibrin gelation. Amyloid plaque and amyloid fiber gel have smaller pores, as revealed by the AFM [10] and TEM images [4] and then smaller Darcy’s permeability coefficient, κ, than the fibrin gel, as shown in the SEM images in the paper.
To quantify the media viscosity change, η, during gelation, we monitored the viscosity change during protein gelation, using a molecular rotor 9-(2-carboxy-2-cyanovinyl)julolidine (CCVJ, Figure 1). The quantum fluorescence yield of CCVJ is dependent on the rotation of two chemical groups around a single bond (Arrow, Figure 1) [12], which are affected by the viscosity of the media. Viscous media restricts the free rotation of the two chemical groups and increases CCVJ’s fluorescence intensity [13,14].

Figure 1: Structure of CCVJ.
The arrow identifies the single carbon-carbon bond responsible for rotation
between the twisted and planar conformations.
Specifically, (1) we examined the effect of fibrin gel on axon’s action potential propagation, (2) quantified the fluid viscosity increase during fibrin gelation, and (3) analyzed the effect of increased viscosity on the axon’s action potential. Results were compared to the presence of lidocaine, a sodium channel blocker. The procedure provided in this paper is the first of such kind in the study of gelation’s effect on action potential and has the potential to become a classical protocol for others to follow.
Materials and Methods
Microviscosity measurements using CCVJ
A viscosity-sensitive fluorescent molecular rotor, 9-(2-Carboxy- 2-cyanovinyl)julolidine (CCVJ; Sigma-Aldrich, St. Louis, MO), was initially dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich) at a stock concentration of 1 mM, and the working solution concentration for CCVJ was 5 μM in 0.1 M phosphate buffer, pH 7.4. Excitation and emission wavelengths for CCVJ are 440 nm and 500 nm, respectively. For all samples and control solutions, 95 μL of the sample was added to 5 μL of 100 μM CCVJ. All measurements were conducted using a microplate reader (BioTek SynergyTM2, Winooski, VT) at room temperature (23.2°C). The gel solutions with no molecular rotor were used as fluorescence blanks and the solutions (no catalyst or substrate) plus the molecular rotor were used as a negative control. To account for small differences in rotor concentration, a normalized fluorescence was calculated as described by Haidekker and Theodorakis [15].
Viscosity change during gelation was determined using either 10 mg/ml or 20 mg/ml fibrinogen and 50 U/ml thrombin at a 10:1 volumeto- volume (v/v) ratio, respectively. As a reference for CCVJ’s response to viscosity changes, fluorescence intensities were measured with CCVJ added to various solutions of glycerol, ranging from 0% glycerol to 95% glycerol (v/v). Glycerol, with no CCVJ added, was used as a blank.
Preparation of sciatic nerves
Xenopus laevis (Xenopus Express, Brooksville, FL) were housed and euthanized according to IACUC approved procedures and accompanying CITI Program training for working with amphibians. Prior to dissection, frogs were placed on ice for 30 min, guillotined, and immediately pithed. Throughout the dissection, amphibian Ringer’s solution (111 mM NaCl, 1.9 mM KCl, 1.06 mM CaCl2, 1.0 mM Trizma base, 5.55 mM glucose, pH 7.6) was used to moisten the dissection area and nerve. The sciatic nerves were then isolated following standard dissection procedures and placed into a dish of Ringer’s solution [16].
Electrophysiology preparation
Isolated sciatic nerves were placed in an iWorx nerve chamber (iWorx, Dover, NH), and glass rods were used to ensure the nerve touched the electrodes of the chamber. A small capsule, created by cutting off the top 80% of a microcentrifuge tube, was placed after the stimulating, but in front of the recording, electrodes. With the NA-100 Neuroamp Extracellular Amplifier (iWorx), the nerve was stimulated at 0.25 V and the CAP was monitored. Once the CAP was stable [17], testing solutions were applied to the capsule and CAP was recorded every 30 s for 15 min. The sciatic nerve was moisturized by applying Ringer’s solution every 5 min, which enables the nerve remained functional for 2-4 h before CAP decline occurred. Without moisturizing, the nerve becomes dysfunctional after 15 min as evidenced by the lack of a CAP after stimulation.
The recorded CAP was analyzed using LabScribe 2 software (iWorx). Fibrin was used as an analog for amyloid plaque and glycerol for a change in local viscosity. Aβ40 was prepared according Stine et al. [18]. In brief, peptide stocks were prepared using 1,1,1,3,3,3-hexafluoro- 2-proponal (HFIP) and stored as 5 mM stocks in dimethyl sulfoxide (DMSO) at -20°C. Oligomeric Aβ40, 100 μM, was prepared using Ham’s F-12, phenol red-free with L-glutamine, cell culture media and incubated at 4°C for 24 h. Fibrillar Aβ40, 100 μM, was prepared in 10 mM HCl and incubated at 37°C for 24 h. Lidocaine (200 mg/ml; Sigma- Aldrich), a sodium channel inhibitor, represented a positive control, while Ringer’s solution represented a negative control.
Extraneural incubations: Gel forming solutions and other reagents were initially applied to the outside of the epineurium membrane, which is known to have pores allowing proteins and small molecules to diffuse across and to act on axons inside the membrane. The fibrin gel was made by mixing a fibrinogen solution (20 mg/mL in 0.9% NaCl, Sigma-Aldrich) with a thrombin solution (50 U/mL in 0.1% BSA, Sigma-Aldrich) in a 10:1 fibrinogen-to-thrombin ratio (v/v) in the incubation chamber (100 μl). This ratio allowed complete gel formation within a minute. In addition, the individual components, fibrinogen and thrombin, were tested separately as controls, and Ringer’s solution was for a baseline. Glycerol (Sigma-Aldrich) was prepared in concentrations of 0% and 50% in 1X Ringer’s solution.
Intraneural incubation: The presence of various solutions and the effect of fibrin gelation inside the epineurium membrane were also tested for effects on the CAP. A 1 ml syringe with a 30 gauge needle was placed into a micromanipulator. Using a dissection scope for visualization, the needle was inserted into the nerve at an angle of 20- 30°. For fibrin, to ensure gelation occurred within the nerve, thrombin was first added to the syringe and the needle was inserted into the nerve. An initial stimulation was recorded and then the fibrinogen was added to the syringe. Immediately, about 20 μl of the mixed solution was injected into the nerve. A longitudinal nerve expansion of about 5 mm was visualized. Controls included Ringer's solution, lidocaine, thrombin and fibrinogen. In addition, the same process was performed using different concentrations of glycerol and Aβ40. Prior to injecting, Aβ40 samples and respective incubation buffers were first diluted to 100 nM using Ringer’s solution.
Fibrinogen-fluorescein isothiocyanate (FITC) conjugation
FITC (Sigma Aldrich), 1 mg/ml in DMSO, and fibrinogen, 20 mg/ ml in 0.1 M sodium carbonate buffer, pH 9.0 were prepared fresh. For every 1 ml of fibrinogen solution, 50 μl of FITC solution was added slowly while stirring and incubated in the dark for 8 h at 4°C. After addition of ammonium chloride, final concentration 50 mM, the mixture was incubated for 2 h at 4°C. Then bromophenol blue and glycerol, final concentrations of 0.1% and 5%, were added to assist with the subsequent separation step. Conjugated fibrinogen (Fg-FITC) was separated using Sephadex G-25 (Sigma Aldrich), eluted with 0.1 M phosphate buffer solution (PBS) buffer, pH 7.4, and stored at 4°C. The protein concentration was determined using Bradford assay. The Fg-FITC concentration was adjusted to 20 mg/ml with non-conjugated fibrinogen.
Confocal imaging of injected fibrin-FITC
The Fg-FITC was mixed in a 10:1 ratio (v/v) with thrombin and injected into the sciatic nerve. After gelation, nerves were washed (3x) in 0.1 M PBS, pH 7.4, for 15 min, transferred to 4% formaldehyde, 0.1 M PBS solution, and incubated for 72 h in the dark at 4°C. After fixation, samples were incubated in 0.1 M PBS for 2 h, followed by 25% sucrose in 0.1 M PBS solution at 4°C for 24 h prior to cryo-sectioning. Nerves were sectioned longitudinally and cross-sectionally at 50 μm with a Leica CM1850 cryotome at -20°C and harvested onto gelatin coated glass slides. The slides were dried overnight at 4°C and stored in a slide box until use.
Gelatin coated slides were prepared by washing in 70% ethanol, drying overnight and dipping 3-5 times for 5 s each in a 0.5% gelatin, 0.05% chromium potassium sulfate solution. Slides were then dried for 48 h and stored at 4°C in slide boxes.
Sectioned samples were rehydrated by two 10 min incubations in 0.1 M PBS, pH 7.4, at 4°C. A molecular tracer, rhodamine B (479.01 g/ mol, 200 μl, 10 μM), prepared in 0.1 M PBS, was applied to the sample twice in the dark, each for 1.5 h. A well was created around the tissue on the slide using nail polish. An additional 200 μl of rhodamine B was then added to the slide before a rectangle glass coverslip was placed onto the well, covering the sample. The slides were then sealed with nail polish, dried, and imaged using confocal microscopy, Nikon Confocal Microscope C1 with EZ-CI software. Images were collected using a Plan Apo 0.45 objective, medium pinhole (60 μm), 1.68 μs pixel dwell, 561 nm laser set to 5% and 488 nm laser set to 10%.
SEM imaging of fibrin and sciatic nerve
Fibrin gel was generated using a 10:1 ratio (v/v) of fibrinogen (20 mg/ml in 0.9% NaCl) and thrombin (50 U/ml in 0.1% BSA, pH 7.4). The fibrin and isolated sciatic nerve were fixed in 3% glutaraldehyde for 72 h, washed in 0.1% PBS, stained in 1% osmium tetroxide/0.1% PBS, dehydrated in increasing concentrations of ethanol and ethyl-acetate, critical point dried using a Denton DCP-1 critical point dryer, and imaged using a JEOL JSM-6380LV scanning electron microscope.
Results
The compound action potential was measured in the presence and absence of fibrin gels formed around the sciatic nerve fibers. The fibrin gel was found to have an inhibitory effect on the propagation of the action potential. Gelation also increases the media viscosity, as indicated by the high fluorescence intensity from the molecular rotors. Media viscosity affects the CAP, which was revealed by an inverse relation between the CAP and the concentration of the glycerol applied extracellularly.
The inhibitory effects of gels on compound action potentials of sciatic nerves
The frog sciatic nerves were stimulated at 0.25 V every 30 s for the generation of CAP. After 2 min of recording for the base level, the fibrinogen and thrombin mixture was applied and the CAP was recorded every 30 s for 15 min.
Fibrin gel formed outside of the porous epineurium membrane around the axon bundles: The nerve fiber’s CAP decreased gradually after fibrinogen, 20 mg/ml, and thrombin mixture was applied outside the nerve (Figures 2A-2C). The CAP decrease occurred mostly in a time window between 120 and 450 s of incubation with the fibrin gel. A rapid decrease of the CAP by 39% ± 12.3 was recorded in the first 450 s. CAP decrease continued thereafter and a reduction by 41% ± 11.5 was recorded at 900 s (n=14) of incubation. Raw CAP traces are displayed in Figure 2A and a plot is presented to provide the mean average of the CAP peak measurement at different time points after application of the fibrinogen and thrombin mixture. The fast and slow phases of CAP decrease appear to correlate with the viscosity increase during fibrin gelation at a compatible timeline (Figure 3B).

Figure 2: Inhibitory effect of fibrin gel on axon��?s CAP.
Frog sciatic nerve CAP (A) was measured with fibrin formed outside (B) and inside (C) of the nerve fiber epineurium membrane. Sciatic nerves were stimulated with
0.25 V. Percentage change of CAP incubated with 20 mg/ml fibrin (☐), Ringerâ�?�?s solution (○), 50 U/ml thrombin (△), 20 mg/ml fibrinogen (▉), and 20% lidocaine (●) are
shown. Lidocaine, a sodium channel inhibitor, was examined as a positive control. Error bars represent �?± SE (*p<0.05)
When applied separately, fibrinogen and thrombin solutions had a small effect on CAP (Figure 2B). Fibrinogen, 20 mg/ml, caused a slight CAP decrease but plateaued after 270 s of incubation and an 8% of CAP reduction (n=4). Thrombin, 50 U/ml, resulted in a slight CAP increase initially and plateaued after 450 s of incubation and a CAP increase by 14.8% ± 3.1 (n=4). As a positive control, lidocaine, 200 mg/ml, a sodium channel blocker [19], reduced the CAP by 90% ± 12.1 after 450 s and 99% ± 1.1 after 900 s (n=2) of incubation. Ringer’s solution had little effect on CAP initially and displayed a slight increase only by 2.9% ± 2.2 (n=10) after 900 s of incubation.
Fibrin gel formed inside the porous epineurium membrane around the axon bundles: The epineurium membrane may reduce the permeation of fibrinogen and thrombin [20,21] and then the amount of fibrin gels formed immediately around the bundles of the nerve fibers. To ensure a better axon encapsulation with fibrin gel, we injected fibrinogen and thrombin mixtures inside the epineurium membrane. Using this method, fibrin decreased the CAP by 70.5% ± 7.6 (n=10) (Figure 2C) over the 900 s of measurement. A decrease of CAP by 35% occurred 30 s after the injection, and then continued at a slower rate through the duration of the measurement. The initial rapid CAP decrease again correlates to the major increase of the media viscosity during fibrin gelation (Figures 2 and 3B).

Figure 3: Media viscosity change during fibrin gelation.
(A) Logarithmic relation between CCVJ fluorescence intensity and the viscosity of 5-85% glycerol. (B) Fluorescence intensity increases during fibrin gelation. The
assay solutions contained 5 μM CCVJ, 20 mg/mL fibrinogen (●) and thrombin (50 U/mL)
The Ringer's solution of equal volume applied inside the porous epineurium membrane had no initial effect and only a slight CAP increase of less than 6% ± 0.8 (n=7) after 900 s of incubation. When applied separately, thrombin and fibrinogen increased the CAP by 15% ± 13.5 (n=4) and 30% ± 12.2 (n=4), respectively. Soluble Aβ40 (100 nM), either as oligomers (n=6) or fibrils (n=6), and respective buffers (n=6) had little effect on the CAP during the 900 s of analysis (Data not shown). As a positive control, lidocaine reduced the CAP to near 0% within the first 30 s of application.
Gelation increases the viscosity of the media
Gels are known to affect the transportation of the media and then the circulation of the ions and molecules around the axons. According to Darcy’s flux, both the Darcy’s permeability coefficient, κ, and the viscosity of the media, η, can affect the value of the Darcy velocity. We employed a molecular rotor, CCVJ, to analyze the viscosity change of the media during fibrin gelation. The fluorescence intensity of CCVJ is related to the viscosity of the media as demonstrated with 5 to 85% of glycerol (Figure 3A). At 5% glycerol, or a viscosity of 1.09 cP, the fluorescence intensity was about 3,312 ± 396 relative fluorescence units (RFU; n=6), at 25% glycerol or 2.22 cP, it was about 7,763 ± 183 RFU (n=6) and at 85% glycerol or 130.71 cP, it was about 67,287 ± 1778 RFU (n=6). When the fluorescence intensity is plotted against the viscosity of the glycerol solution (Figure 3A), an empirical formula was derived:

where I is the intensity in RFU and η is the dynamic viscosity in cP. A logarithmic linear increase of CCVJ fluorescence intensity exists, as predicted by the Forster-Hoffman [15].
The media viscosity increases during fibrin gelation as evidenced by the CCVJ fluorescence increase (Figure 3B). As fibrinogen, 20 mg/ mL (n=6), polymerized into a fibrin network, the fluorescence of CCVJ increased exponentially. About 75% of the peak intensity was reached in 60 s, followed by a slow increase to the maximum intensity after 480 s. The peak fluorescence intensity, reached about 300 s after mixing, was about 1.25-fold higher than that measured immediately after mixing of the fibrinogen and thrombin solutions. Using the equation obtained from the analysis of glycerol, a viscosity change of fibrin gelation can be derived:
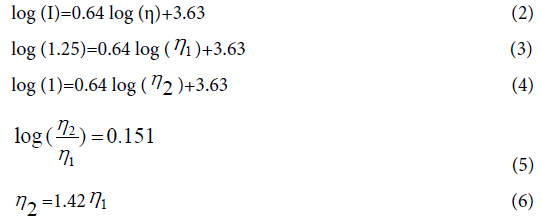
This relative fluorescence change from 1 to 1.25 corresponds to an estimated viscosity increase of 1.42 fold when fibrinogen solution becomes fibrin. The actual fluorescence increase of fibrin gelation might be larger than 1.25 fold if light scattering effect is considered for both absorption and emission.
CAP is dependent on extracellular viscosity
Darcy’s flux velocity, which determines the circulation rate of ions and molecules around axons, is dependent on viscosity. To test whether and how CAP is dependent on extracellular viscosity, we analyzed the effect of glycerol on the sciatic nerve’s action potential. Glycerol was applied to the nerves both outside and inside of the epineurium membrane. Extra-epineurium incubations of glycerol caused a continuous decrease of the CAP during the entire time of 900 s of analysis. A 36% ± 15.1 decrease of CAP was recorded for 100% (n=5) and a 9% ± 2.8 decrease for 50% glycerol (n=7) after 900 s of observation (Figure 4A). Conversely, compared to extra-epineurium, intra-epineurium application of glycerol reduced CAP to a much greater extent (Figure 4B).Glycerol at 50% (n=4) reduced CAP by 40% within the first 15 s and by 60% after 120 s. Glycerol at 100% reduced the CAP by 96.5% ± 1.5 and glycerol at 50% reduced the CAP by 79.2% ± 7.9 after 900 s of incubation.

Figure 4: Inhibitory effect of glycerol on axon’s CAP. Frog sciatic nerve CAP was measured with glycerol present outside (A) and inside (B) of the nerve fiber’s epinerium membrane. Sciatic nerves were stimulated with 0.25 V. Percentage change of CAP incubated with Ringer’s solution (○), 50% glycerol (Δ) or 100% glycerol (▉) is presented. Error bars represent ± SE (*p<0.05).
Imaging fibrin gel and the sciatic nerve fiber
Scanning electron micrograph images of fibrin (Figure 5A) and the sciatic nerve (Figure 5B) reveal the structural characteristics of each sample. Under our condition, fibrin, 20 mg/ml, forms a dense network of fibers with varying pore diameters up to 1 μm. The sciatic nerve consists of a single bundle of fibers surrounded by the epineurium (Figure 5B). A single nerve, 400-1000 μm in diameter, contains thousands of myelinated axons, 5-20 μm in diameter. Cross and longitudinal sections reveal little extracellular space and a loose endoneurium collagen matrix (Data not shown).

Figure 5: Structure of fibrin gel and the sciatic nerve.
Scanning electron micrograph of fibrin (A), frog sciatic nerve (B) and confocal image of fibrin gel formed inside the epineurium membrane (C) are shown. Fibrin
gel was generated using a 10:1 ratio (v/v) of fibrinogen (20 mg/ml in 0.9% NaCl) and thrombin (50 U/ml in 0.1% BSA, pH 7.4). The sciatic nerve was fixed in 3%
glutaraldehyde, washed in 0.1% PBS, stained in 1% osmium tetroxide/0.1% PBS, dehydrated in increasing concentrations of ethanol and ethyl-acetate, dried and
imaged using the SEM. FITC-labeled fibrinogen and thrombin mixture was injected inside the epineurium membrane of the sciatic nerve (C)
Confocal imaging of longitudinal sciatic nerve (Figure 5C) reveals that not all sciatic nerve axons were encapsulated by fibrin. Using FITCFigure fibrinogen, we found that the fibrinogen and thrombin mixture was successfully applied inside the epinerium membrane. The fibrin-FITC appears green and was found longitudinally along the axons. The partial CAP reduction in the presence of fibrin gel is due to the incomplete encapsulation of all the axons by fibrin gel. Some axons are fibrin free and may remain conductive to the action potential.
Discussion
What causes neuronal death in AD remains unknown. The discovery that amyloid fibers can further aggregate and form gels and the understanding that gels are capable of restricting bulk flow led us to test, for the first time, the new hypothesis that amyloid plaques as protein gels can inhibit action potential propagation. In this paper, we found that fibrin gels formed outside the frog sciatic nerve inhibits the CAP. Fibrin gelation increased the media viscosity. CAP was also reduced when glycerol was applied extracellularly to the nerve fibers. The results suggest that amyloid plaques may inhibit the CAP of the nerve fibers, which may trigger a cascade of reactions leading to the eventual neuronal death in AD.
According to Darcy’s law, increased media viscosity, μ, as observed in gelation, would result in a proportionally flux velocity reduction of the fluid moving through the gel. In a previous paper we provided evidence that amyloid plaques are gels with pore sizes of tens of nanometers [10], which are much smaller than micron-sized fibrin pores. In another words, a higher media viscosity and smaller Darcy’s permeability coefficient are expected for amyloid plaque than that for the fibrin gel, which would result in a reduced fluid flux velocity through amyloid plaque than that through fibrin gel. Amyloid plaque as a protein gel would be more effective than the fibrin gel in restricting fluid flow. If the inhibition of the action potential propagation is a result of reduced circulation of fluid and then ions and molecules around the axons, one would expect a larger reduction of the CAP for nerve fibers wrapped by amyloid plaque than by fibrin gel. It makes sense that amyloid plaque in AD may kill neurons because of its initial inhibition of the nerve’s action potential.
Neurites are vulnerable to suffocation by the protein gels or amyloid plaques due to their narrow shape and a long travelling distance. Action potential propagation requires free access of interstitial ions and oxygen to neurites. Such an access would be attenuated by the amyloid plaques wrapping around the neurites. Inhibition of action potential propagation could paralyze a synapse’s function and trigger immunological destruction of the disused synapses by microglia and astrocytes [22-24]. Massive synapse loss is a hallmark of AD. The denied access model introduced in this paper provides a link between amyloid plaques, neurite pathology, synaptic loss, and microglia and astrocyte phagocytosis.
The effect of glycerol on CAP provides additional evidence supporting the idea that limiting ion or molecular motion inhibits action potential propagation. Viscosity of glycerol increases proportionally with concentration, and the diffusion coefficient is inversely proportional to viscosity, based on Stokes-Einstein-Sutherland relationship. In addition, the rate of bulk flow is negatively affected by the friction force, and the latter is proportional to the viscosity of the solution. Thus, both bulk flow and diffusion are, in theory, negatively affected by the increased glycerol concentration.
Based on above analysis, we introduce the denied access model (Figure 6). When surrounded by amyloid plaques/protein gel, neurites are denied to have free access to interstitial ions, nutrients, signaling molecules, and metabolic waste drainage pathways, which results in the inhibition of the compound action potential propagation. This is an alternative model for the AD community to consider, although continued investigations are required to verify the model and to fulfill the details.

Figure 6: Denied access model.
The neurite regions surrounded by amyloid gel plaques are denied from accessing to nutrient and signaling molecules as well as to the drainage pathways for
metabolic wastes. This denied access leads to the inhibition of the action potential propagation and eventually the degeneration of the disused neurites.
The denied access model is simple and inclusive of the physics and engineering perspective of gels or plaques, in addition to the biochemical nature. The inclusion of physical properties such as bulk flow and diffusion is seen in the illustration of atherosclerosis, thrombosis, gallbladder stones, and kidney stones, where aggregation of biomolecules, large and small, is also an early event of disease pathogenesis. The transportation phenomenon, or bulk flow and diffusion, through gels are an active research area in engineering.
Conclusion
According to the proposed model, individual amyloid fibers would be nontoxic since they do not suffocate the neurites. Tissues are full of different kinds of protein fibers. Amyloid fibers are unique due to their resistance to protease digestion while most other fibers in vivo such as collagen can be degraded. Oligomers are claimed by some as the neuronal pathogen. Others are concerned about the lack of a common mechanism for how these oligomers exercise their toxicity [7]. The claim of toxic oligomers is focused on the chemical nature of the protein aggregates without adequate consideration of the physics and the physical presence of plaques. These spheres are diverse with no defined structure, and thus should be correctly called colloids [25,26]. Not calling these spheres colloids also detaches amyloid research from colloidal science, a large area of physical science that has accumulated a large body of knowledge over the last one and half centuries. Similarly, not calling the amyloid plaque a gel also detaches AD research from the knowledge accumulated in engineering on transportation phenomena in gels and porous materials. The name "oligomer" is a misnomer. An oligomer, by definition, is a molecule that consists of a relatively small and specifiable number of monomers, usually less than five (see the Free Dictionary).
Much investigation is required for the details of the denied access model including the mechanism leading to CAP reduction. In addition to ions and small molecules, accessibility to proteins such as insulin would be impaired as well.
Author Contributions
Dauberman and Briet performed the molecular rotor and electrophysiology experiments. Dauberman completed FITC conjugation and SEM imaging. Xu proposed the model, and designed the overall experimental approach to test the model. Dauberman and Xu wrote the manuscript. All authors contributed to data analysis.
Acknowledgement
The study was supported by NASA, The Florida Space Grant Consortium, and The Community Foundation for Brevard. We thank Dr. David Tipton and Dr. Daniel Woodard of Kennedy Space Center for their support. We also thank Dr. Michael Grace for the use of his cryotome, Dr. David Carroll for frog husbandry assistance and the use of a micromanipulator and the Florida Institute of Technology microscopy center.
References
- Xu S (2007) Aggregation drives "misfolding" in protein amyloid fiber formation. Amyloid 14: 119-131.
- Tan SY, Pepys MB (1994) Amyloidosis. Histopathology 25: 403-414.
- Wisniewski HM, Vorbrodt AW, Wegiel J, Morys J, Lossinsky AS (1990) Ultrastructure of the cells forming amyloid fibers in Alzheimer disease and scrapie. Am J Med Genet Suppl 7: 287-297.
- Wisniewski HM, Wegiel J (1995) The neuropathology of Alzheimer's disease. Neuroimag Clin N Am 5: 45-57.
- Yamaguchi H, Nakazato Y, Shoji M, Ihara Y, Hirai S (1990) Ultrastructure of the neuropil threads in the Alzheimer brain: Their dendritic origin and accumulation in the senile plaques. Acta Neuropathol 80: 368-374.
- Dickson DW (1997) The pathogenesis of senile plaques. J Neuropathol Exp Neurol 56: 321-339.
- Benilova I, Karran E, De Strooper B (2012) The toxic A�?�?�?² oligomer and Alzheimer's disease: An emperor in need of clothes. Nat Neurosci 15: 349-357.
- Hardy J (2009) The amyloid hypothesis for Alzheimer's disease: A critical reappraisal. J Neurochem 110: 1129-1134.
- Burnett LC, Burnett BJ, Li B, Durrance TD, Xu S (2014) A lysozyme concentration, pH and time-dependent isothermal transformation diagram reveals fibrous amyloid and non-fibrous, amorphous aggregate species. Open Journal of Biophysics 4: 39-50.
- Woodard D, Bell D, Tipton D, Durrance S, Burnett LC, et al. (2014) Gel formation in protein amyloid aggregation: A physical mechanism for cytotoxicity. PLoS ONE 9: e94789.
- Morse RP, Evans EF (2003) The sciatic nerve of the toad Xenopus laevis as a physiological model of the human cochlear nerve. Hear Res 182: 97-118.
- Haidekker MA, Theodorakis EA (2010) Environment-sensitive behavior of fluorescent molecular rotors. J Biol Eng 4: 11.
- Akers WJ, Cupps JM, Haidekker MA (2005) Interaction of fluorescent molecular rotors with blood plasma proteins. Biorheology 42: 335-344.
- Grabowski ZR, Rotkiewicz K, Rettig W (2003) Structural changes accompanying intramolecular electron transfer: Focus on twisted intramolecular charge-transfer states and structures. Chem Rev 103: 3899-4032.
- Haidekker MA, Theodorakis EA (2007) Molecular rotors--fluorescent biosensors for viscosity and flow. Org Biomol Chem 5: 1669-1678.
- Katsuki R, Fujita T, Koga A, Liu T, Nakatsuka T, et al. (2006) Tramadol, but not its major metabolite (mono-O-demethyl tramadol) depresses compound action potentials in frog sciatic nerves. Br J Pharmacol 149: 319-327.
- Benzon HT, Gissen AJ, Strichartz GR, Avram MJ, Covino BG (1997) The effect of polyethylene glycol on mammalian nerve impulses. Anesth Analg 66: 553-559.
- Stine WB, Jungbauer L, Yu C, LaDu MJ (2011) Preparing synthetic A�?�? in different aggregation states. Methods Mol Biol 670: 13-32.
- Hondeghem LM, Katzung BG (1977) Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta 472: 373-398.
- Oldfors A (1981) Permeability of the perineurium of small nerve fascicles: An ultrastructural study using ferritin in rats. Neuropathol Appl Neurobiol 7: 183-194.
- Schwarz JR, Ulbricht W, Wagner HH (1973) The rate of action of tetrodotoxin on myelinated nerve fibres of Xenopus laevis and Rana esculenta. J Physiol 233: 167-194.
- Chung WS, Welsh CA, Barres BA, Stevens B (2015) Do glia drive synaptic and cognitive impairment in disease? Nat Neurosci 18: 1539-1545.
- Miyamoto A, Wake H, Moorhouse AJ, Nabekura J (2013) Microglia and synapse interactions: Fine tuning neural circuits and candidate molecules. Front Cell Neurosci 7: 70.
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, et al. (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74: 691-705.
- Xu S, Bevis B, Arnsdorf MF (2001) The assembly of amyloidogenic yeast sup35 as assessed by scanning (atomic) force microscopy: An analogy to linear colloidal aggregation? Biophys J 81: 446-454.
- Xu S (2009) Cross-beta-sheet structure in amyloid fiber formation. J Phys Chem B 113: 12447-12455.
Relevant Topics
- Advanced Parkinson Treatment
- Advances in Alzheimers Therapy
- Alzheimers Medicine
- Alzheimers Products & Market Analysis
- Alzheimers Symptoms
- Degenerative Disorders
- Diagnostic Alzheimer
- Parkinson
- Parkinsonism Diagnosis
- Parkinsonism Gene Therapy
- Parkinsonism Stages and Treatment
- Stem cell Treatment Parkinson
Recommended Journals
Article Tools
Article Usage
- Total views: 3583
- [From(publication date):
August-2017 - Aug 16, 2025] - Breakdown by view type
- HTML page views : 2719
- PDF downloads : 864