Stochastic Considerations into the Origins of Sporadic Adult Onset Neurodegenerative Disorders
Received: 19-Jul-2019 / Accepted Date: 05-Aug-2019 / Published Date: 12-Aug-2019 DOI: 10.4172/2161-0460.1000473
Abstract
Objective: Alzheimer’s disease, Parkinson’s disease, frontotemporal dementia and other neurodegenerative disorders share common properties including protein interactions, cellular reactions, inflammatory process involving microglia, prion-like propagation in a neuronal network, synaptic and neuronal loss. The misfolding and aggregation of specific proteins seems to be an early and obligatory event of which the antecedents are unknown.
Methods: Studies in prion diseases and AD implicate the conversion of disease-specific proteins into aggregates of prion-like beta-sheets. Most of the common neurodegenerative disorders are sporadic, with <5% resulting from genetic mutations. This work aims to explain the mechanisms by which most neurodegenerative disorders are sporadic.
Results: It is posited that variation in protein sequences may be caused by stochastic processes at a DNA, mRNA or protein level. This sequence variation is resistant to the neuron’s normal control mechanisms and results in disease through protein misfolding, over-proliferation and spread. If not handled by the cell’s normal mechanisms, such as phagosome function, the process might result in disease.
Conclusion: The association with neurodegenerative disorders with age correlates with failure of the cell’s normal mechanisms, such as autophagosomes and agressomes, to deal with this sequence variation. These considerations raise evolutionary questions as to the origins of neurodegenerative disorders in humans.
Keywords: Stochasticity; Neurodegenerative disorders; Prion diseases; Protein interactions; Alzheimer’s disease; Adult onset
Introduction
Every day in my clinical work I ask the question: Why do young adults develop sporadic Alzheimer’s disease (AD), Parkinson’s disease (PD) and other neurodegenerative disorders? This work attempts to set out the reasons as to why this might happen in the absence of genetic mutations.
The pathology of AD, PD, Frontotemporal Dementia (FTD), Motor Neuron Disease (MND) and prion diseases share common properties including protein-protein interactions, cellular reactions involving microglia, inflammatory processes, prion-like propagation through a neuronal network, resulting in synaptic and neuronal loss. The misfolding and aggregation of specific proteins seems to be an early and obligatory event in all of these disorders of which the antecedents are unknown. Studies in prion diseases and AD implicate the conversion of disease specific proteins into aggregates of prion-like beta-sheets as a fundamental process. It appears that prion-like corrupted protein templates are a feature of these neurodegenerative disorders. Misfolding, aggregation, trafficking and pathogenicity of the involved proteins are fundamental mechanisms shared by the common neurodegenerative disorders, and are responsible for significant global burden of disease and costs [1,2]. It is therefore essential to understand this process.
AD may present at different ages. AD may be young onset- that is, begins before the age of 65 years- or old onset- starting after 65 [3,4]. Furthermore, AD can present in different ways. For example, there is the amnestic presentation, in which patients develop a difficulty with episodic memory, implying mesial temporal structure involvement, which in time spreads throughout the brain. Other presentations include a linguistic presentation in which speech is primarily affected and known as a logopenic variant of AD; posterior cortical atrophy, a variant in which patients develop visuospatial dysfunction on account of initial involvement of the parieto-occipital areas of the brain; and a frontal variant, in which patients have predominant executive dysfunction from frontal involvement [5].
How does one explain the differences in age of onset? How do does one account for the spectrum of clinical presentations given our current knowledge of the pathomechanisms of AD in the light of the misfolding and aggregation of specific proteins?
In our studies of AD, we believe that there are differences between young and old onset disease. In our research, we have found that most young-onset AD patients do not carry mutations associated with familial AD [6], an observation shared by others [7,8]. Less than 1% of our cohort with AD had a mutation in the Amyloid Precursor Protein gene (APP) or presenilin-1 or 2. Also, most late-onset AD patients do not carry mutations. They might carry the APOE-4 genotype, which seems to predispose to amyloidosis. If assumed that most familial AD is not due to genetic mutations, how does one then explain the development of AD through the life spectrum? How does one explain the sporadic burden of this disorder which, throughout the 21st century, has become a global public health problem? Furthermore, how does one account for differences in presentation in AD, and in particular young-onset AD, in terms of a typical amnestic presentation, posterior cortical atrophy, linguistic presentations and frontal variants? How, in the modern era, does one account for these observations? Additionally, how does one explain these differences given our understanding of misfolding and aggregation of specific proteins in the absence of gene mutations? If the gene mutation hypothesis only accounts for the minority of AD, then what is the fundamental process driving the remainder of AD? These considerations not only apply to AD, but are relevant to prion diseases in their different syndromic presentations; and are also relevant to PD, MND and FTD. What is it that drives the fundamental processes converting a healthy brain into a dementing brain secondary to the processes of misfolding, aggregation, propagation through a neuronal network with synaptic and neuronal loss?
AD, PD, prion diseases, MND and FTD are sporadic in the majority, and not related to gene mutations [7,9-14]. Could it be that the fundamental process driving the origins of these neurodegenerative disorders is stochastic?—that is, fundamental variation in the sequence of key proteins or other proteomic changes: prion proteins in Creutzfeld Jakob Disease (CJD), tau and Aβ peptides in AD, α-synuclein in PD, a number of proteins in MND and FTD (C9orf72, Tau on PGRN), all leading to devastating consequences of inexorably progressive neurodegenerative diseases. It is speculated that random variation in protein sequences (or other proteomic divergence) of key proteins is a fundamental process. Stochastic events in brain protein synthesis may be physiological and essential and would be predicted in certain circumstances, such as storage and transmission of information [15]. Deviations in protein sequences are probably part of normal brain function which, in some individuals results in neurodegeneration. It is this concept that is being developed in this research; that is, the stochastic principles underlying synthesis of certain proteins in the brain, which ignite the neurodegenerative flame.
Support for this concept comes from the studies of Qiang et al. where an investigation of amyloid fibrils studied in vitro suggested that variations in the Aβ fibril structure may correlate with AD phenotype, similar to the observations in prion diseases [16]. These authors use solid state nuclear magnetic resonance measurements on Aβ40 and Aβ42 fibrils prepared by seeded growth from extracts of AD brain cortex. These authors compared two atypical AD subtypes, the rapidly progressive form, the posterior cortical atrophy variant and the typical prolonged duration amnestic form. They observed that a single Aβ40 fibril structure was mostly found in samples from typical AD and posterior cortical atrophy, whereas Aβ40 fibrils from rapid AD had a significant proportion of additional structures [16]. The finding suggested that there is structural heterogeneity for all subjects with two prevalent structures, signifying the existence of a specific predominant Aβ40 fibril in typical and posterior cortical atrophy AD, with additional structures in rapid AD suggesting that there are qualitative differences in Aβ40 and Aβ42 aggregates in the brain tissues of patients with AD. It is posited that these structural differences are determined by the amino acid sequence and other protein structural abnormalities in these fibrils, which is the origin of these stochastic mechanisms. For AD, where there is interaction between two proteins Aβ, the fundamental protein of the neuritic plaque versus microtubule associated protein tau, which in its phosphorylated form makes up the neurofibrillary tangles. There is debate on the role of these two proteins in the pathophysiology of AD. Tau aggregation in AD correlates better with memory and spread suggesting that neuritic plaques may be bystanders, perhaps even protective, as some studies suggest, and the driving process may be tau or some other factor operational on tau- that is, the fundamental defect is in tau amino acid sequences, determined by stochastic events, which drives AD [17,18]. The implication is that some protein sequences are more aggressive than others is supported by the studies of Qiang et al. leading to rapidly progressive Alzheimer syndromes [16]. It is also suggested that this sequence variation may initially occur in different neuroanatomical regions of the brain, such as in the parieto-occipital areas, in posterior cortical atrophy versus the typical amnestic version from mesial temporal involvement, raising the suggestion that frontal variants and linguistic variants of AD are driven by stochastic events in space, time and sequence, which eventually result in a progressive dementing disease as a result of protein spread. Such stochastic considerations also power other neurodegenerative conditions.
This research aims to develop this concept of the importance of stochastic operations in the genesis of neurodegenerative diseases.
Stochastic Effects
It has been postulated that after injury stochastic changes take place and lead to damage and reconstruction which can, in turn, induce cellular changes. That is, certain microenvironmental influences like pH affect the sequences of the key proteins in neurodegeneration, leading to certain primary structures that favour misfolding and resistance to proteosome function [19]. During normal cell metabolism, each cell undergoes a large number of biochemical reactions that require a diversity of biological molecules. Under the constraints of space, resources and the need for metabolic conservation, cells optimise resources because of low stock and high flux. As a result, although the types of molecules might be large and diverse, only a few copies of each molecule are involved. In addition, the biochemical reactions that occur at the scale of a single cell are probably influenced by stochastic effects, so that they cannot be predicted because of high intrinsic noise. Furthermore, cell metabolism is affected by dynamic physiological activities such as fluctuations in metabolism, transcription factors, hormones and other variables. This further increases the extent of uncertainty in biochemical reactions (extrinsic noise). Stochastic fluctuations in the physiological microenvironment in cells also directly influence cellular functions, resulting in cellular diversity within the same tissue or organ. The stochastic effects of metabolic regulatory networks and associated biochemical reactions probably promote population diversity. This allows large cell populations to cope with stress in the external environment and thus confers evolutionary advantages. A single transgenesis event is unlikely to produce the same protein products because of the stochasticity of protein expression and the combination of expression products. In turn, stochasticity of gene expression affects biochemical processes. The copious number of proteins fluctuates randomly, producing perturbation and downstream biochemical reactions. Therefore, even cells carrying exactly the same genes or cloned in the same environment may present with phenotypic diversity [20]. Stochastic effects are observed in the DNA replication process, which involves the use of restriction enzymes to correct mismatch [21,22].
The stochasticity of biochemical reactions likely becomes more pronounced with aging. In the middle stage of the cell cycle the restorative synthesis and allocation of molecules increases. Variation between adjacent cells is also enhanced. The increased stochasticity of DNA replication aggravates DNA injury and mutation- the incidence of spontaneous mutation is greater than expected. Pathological problems such as mitochondrial DNA diseases and other lesions such as tumours can arise from such mutations. The stochasticity of biochemical reactions may cause cells to become non-viable despite a young and energetic body. During aging, physiological and pathological changes can accumulate, thereby complicating stochastic change and protein expression. For example, diabetes in middle age and elderly individuals probably intensifies non-enzymatic glycosylation occurring in the human body. Thus, functional decline in proteins facilitates aging.
Randomness in amino acid sequences occurs by stochastic processes and natural selection will eliminate unsuitable sequences [23]. How does microevolution coordinate the macroevolution and how does natural selection play a role in this process? Stochastic aspects of biochemical reactions create the possibility of changes in cellular elements; by contrast, mechanical and biochemical loads provide the direction for such changes. Biochemical reactions have intrinsic randomness in the reproduction of molecules. Natural selection not only eliminates unsuitable traits, but also guides the formation of new and favourable characteristics.
Stochasticity and biochemical reactions
Protein molecules can be modified by intracellular microenvironments, such as oxidation of cellular amino acid pools. The changes in biochemical environments also enhance the stochasticity of biochemical reactions and reduce the accuracy of mRNA translation resulting in mistranslation of the genetic code. The proteins can be selectively damaged, degraded or metabolised. Then, in newly synthesised proteins, the site of one amino acid could be occupied by another- substitution of Leu by Ser- which might lead to an adaptive change in the structure and function of proteins [24].
In mammals and birds there is GC bias which can be interpreted as a GC-bias mismatch repairing trend- that is, using G or C as the same template- in the mismatch repair process. Randomness causes uncertainty in the development and repair at a single cell level because microenvironments for cellular biochemical reactions cannot be the same. This is possible because a closely coordinated development and repair mechanism is established in the chaotic interaction between biomolecules. Changes in the macroenvironment can cause rapid alterations in the physiological factors of the internal environment (biochemical load). Such changes may further affect development, growth or repair which transforms the form, structure and function of tissues and organs; as a result, this process drives the adaptive evolution of species [25-33].
Furthermore, stochasticity can be influenced by random biochemical reactions with interaction among biochemical reactions and molecules.
Currently, evolutionary mechanisms such as transformation and diversification remain poorly understood [34]. A relationship between the forces of macroevolutionary and microevolutionary processes needs to be understood. The link between natural selection and change in biological adaptation is important, particularly the mechanism by which new structures and organs are formed.
Evidence has shown a significant role of natural selection at the molecular evolutionary levels. Molecular mutations are manifested as either favourable mutations or unfavourable mutations under different environmental conditions. During the critical stages of species formation, DNA molecules are under positive selection pressure, resulting in sharp increase in the replacement rate of basic groups and corresponding genomic adjustment. Thus, organisms generate a series of microevolutions to affect macroevolution [35].
The protein product encoded by one gene often regulates that of other genes; the time delay and other variables controlling transcript initiation and translation has been studied. Simulation of the processes of gene expression shows that proteins are produced from an activated promoter in short bursts of variable numbers of proteins that occur at random time intervals. As a result, there can be significant differences in the time between successive events in regulatory cascades across a cell population. In addition, the random pattern of expression of competitive effectors can produce probabilistic outcomes in switching mechanisms that select between alternate regulatory paths. The result can be a partitioning of the cell population into different phenotypes as the cells follow different paths. There are numerous examples of phenotypic variations in isogenic populations of both prokaryotic and eukaryotic cells that may be the result of these stochastic gene expression mechanisms [36].
Elowitz et al. examined clonal populations of cells to explain why phenotypic variation is found [20]. Such heterogeneity can be essential for many biological processes and is conjectured to arise from stochasticity, or noise in gene expression. These authors constructed strains of E. coli that enabled detection of noise and discrimination between the two mechanisms by which it is generated. Both stochasticity inherent in the biochemical process of gene expression “intrinsic noise” and fluctuations in other cellular components “extrinsic noise” contribute substantially to overall variation- that is, there are two processes, the stochasticity within the biochemical process itself and other variations in the cellular microenvironment (ie, the biophysics, which might influence this variation in stochasticity and outcome). Transcription rate, regulatory dynamics and genetic factors control the amplitude of noise. These authors helped establish a quantitative foundation for modelling noise in genetic networks and reveal how low intracellular copy numbers of molecules can fundamentally limit the precision of gene regulation. Martin developed the concept that stochasticity modulates patterns of ageing and is influenced by the constitutional genome, both processes driving the pace and patterns of ageing [37]. Stochastic processes are probably major players in large intra-specific variations in life-span and health-span. Chance events result in somatic mutations and protein synthesis errors which might cause a neurodegenerative condition, other variations in gene expression and epigenetic effects may allow other stochastic effects on aging. Kosik et al. highlighted the importance of mRNAs in post-transcriptional gene regulation and are abundant in the central nervous system [38]. These mRNAs might be relevant to neuronal plasticity and probably contribute to stochastic mechanisms underlying neurodegeneration. There are highly conserved pathways of mRNA biogenesis, closely linked to the transcription and translation of mRNAs; there are 500 known human mRNA sequences, of which 21 nucleotides bind to multiple mRNA targets, and are important in the regulation of gene expression and add another layer of stochastic influence. Rahman et al. developed a stochastic model to compute in vivo protein turnover rate using isotope labelling and high-throughput liquid mass spectrometry, and demonstrated a stochastic process with Gaussian and an Ornstein-Uhlenbeck covariance matrix [39]. As people age their protein synthesis rate is less. If certain mutations are depleted, the lifespan of the organism is increased. mRNA translation is energetically demanding, utilising 50% of the total energy in the cell. A reduction of protein synthesis probably is the bioenergetic basis underlying the cells inability to deal with stochastic production of renegade proteins and contributing to variations in ageing and neurodegeneration [40]. In 2002, Dean suggested that recombination and the effect of stochastic protein synthesis is controlled by complex feedback mechanisms between genes and the larger biological environments leading to uncertainty of outcomes [41].
Stochasticity and Genes
Elowitz et al. studied stochastic gene expression in single cells and found that clonal populations of cells have phenotypic variability; such heterogeneity may be essential for biological processes and arises from stochastic or noise changes in gene expression [20]. The authors looked at two strains of E. coli and found that there was intrinsic and extrinsic noise and that these led to variation in the control of the amplitude of the noise, to transcription rate to regulatory dynamics, and further genetic factors. This resulted in a quantitative foundation for modelling noise in genetic networks and low intracellular copies of molecules can limit the precision of gene regulation [20]. Rodriguez et al. showed that DNA sequence evolution through nucleotide substitution is a stationary Markov process. Four models with 6 independent promoters were studied and showed that nucleotide substitution rates and variations in protein sequence might be based on stochastic models [42].
McAdams & Arkin addressed the question that gene activity is controlled by molecular signals and determine when and how genes are transcribed [35]. In genetically controlled pathways, protein production of one gene regulates the expression of other genes. There are time delays from the first promoter by increased concentration that affect the next promoter and the protein accumulates. Proteins are produced from an activated promoter in short bursts of variable numbers at random time intervals. Large differences occur in the time between successive events in regulatory cascades across a cell population. Such random patterns of expression of competitive events have a probabilistic outcome in switching mechanisms that selects between alternative regulatory pathways. This might give rise to alternative cellular populations with different phenotypes following different pathways, which results in phenotypic variation in isogenic populations and points to stochastic gene expression mechanisms [35].
The emergence of the human brain is one of the evolution’s most compelling mysteries. With its singular importance and astounding complexity, understanding the forces that gave rise to it is a major undertaking. Recently, the identification and publication of the complete genomic sequence of humans, mice, chimpanzees and macaques has allowed considerations of the genetic substrates of natural selection. It was suggested that these molecular events are both at a genetic and protein sequence level [43].
Somatic mutations as determined by Single Nucleotide Variants (SNVs) occur in the central nervous system especially in embryogenesis but continue, although less so, into adulthood [44]. About 1500 SNVs might be found in neurons from the human neocortex [45]. These SNVs are determined stochastically and, if in the appropriate genesproteins set necessary for neurodegeneration, might lead to disease. It is now possible to detect the emergence of mutations in single brain cells during development [46]. Whole-exome sequencing of postmortem hippocampal neurons discovered SNVs in AD brains that increased with age; interestingly, about one-fifth of these SNVs in AD originated in genes implicated in the hyperphosphorylation of tau- the molecular basis of neurofibrillary tangles and one of the stamps of AD [47]. We submit that somatic neuronal mutagenesis in certain genes in vulnerable adults is a contributing factor to the genesis of sporadic neurodegenerative disorders.
Gene expression is a mechanism by which proteins are synthesised using amino acids. Stochastic processes have been emphasised in that they might influence the time it takes for the protein to be assembled [48]. These authors found that binding times vary and that protein synthesis fluctuates, so does the need for protein translation. Stochastic processes affecting protein synthesis are governed by complex feedback mechanisms between genes and other molecular influences leading to variation in amino acid sequence and function [49].
mRNA translation and protein synthesis is a major component for transformation of the genetic code into cellular activity. This complicated, multi-step process is divided into three phases: initiation, elongation and termination. Initiation is a step at which the ribosome is recruited to the mRNA and is regarded as the major rate limiting step in translation, while elongation consists of elongation of a polypeptide chain; both steps are frequent targets for regulation, which is defined as a change in the rate of translation of an mRNA per unit time (Figure 1). In normal brain, translation is a key mechanism for regulation of memory and synaptic plasticity consolidation. That is, the offline processing of required information. These regulatory processes may differ between different brain structures or neuronal populations. Moreover, dysregulation of translation leads to pathological brain function such as memory impairment. Both normal and abnormal function of the translation machinery is believed to lead to translation up-regulation or downregulation of a subset of mRNAs. However, the identification of newly synthesised proteins and the determination of rates of protein synthesis, with degradation taking place in different neuronal types and cellular compartments at different time points in the brain, demand new proteomic methods and system biological approaches [50].
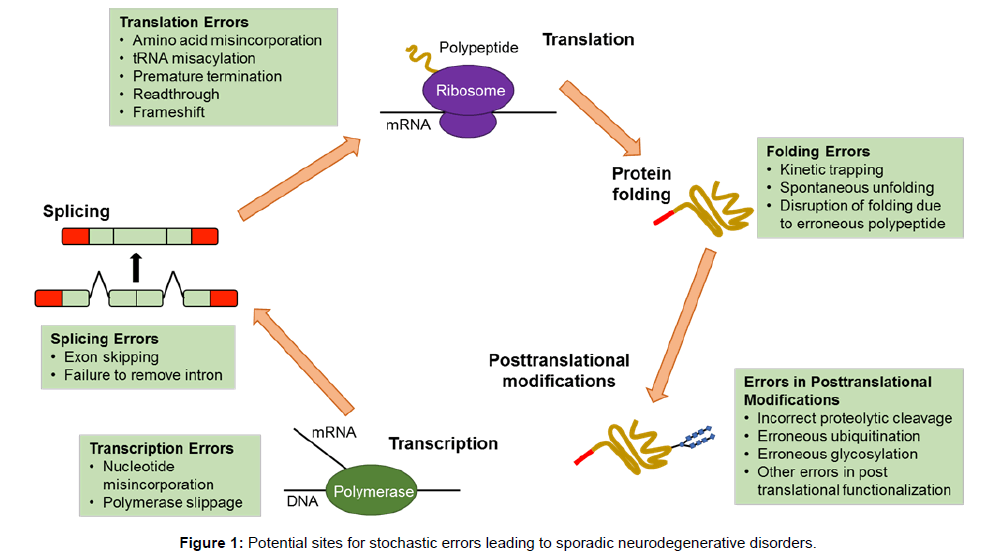
Figure 1: Potential sites for stochastic errors leading to sporadic neurodegenerative disorders.
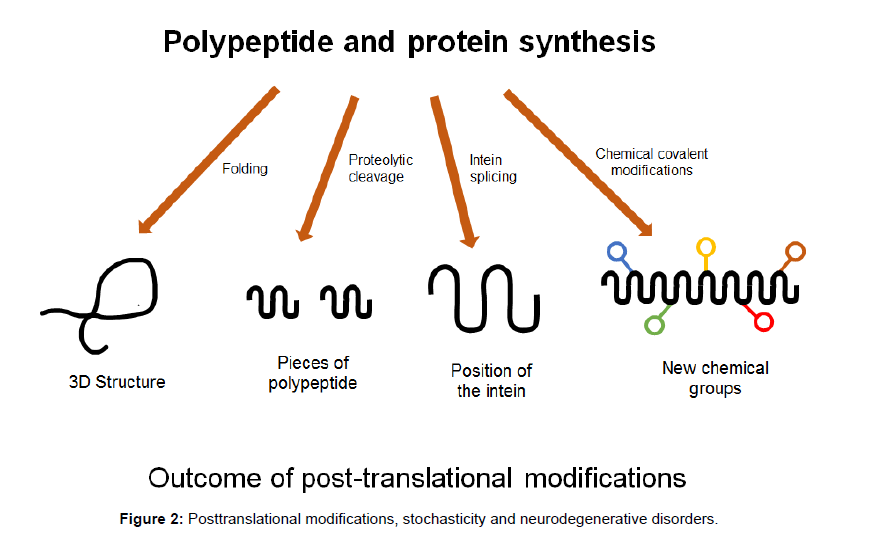
Figure 2: Posttranslational modifications, stochasticity and neurodegenerative disorders.
Newly synthesised chains of amino acids transform themselves into perfectly folded proteins which depend on the intrinsic properties of the amino acid sequence and on multiple contributing influences from the crowded cellular milieu and are modified by post-translational modifications which must be governed by stochastic principles (Figures 1 and 2). Folding and unfolding are crucial ways of regulating biological activity and targeting proteins to different cellular locations. Aggregation of misfolded proteins that escape the cellular quality control mechanisms is a feature of the neurodegenerative disorders under consideration. Many newly synthesised proteins are translocated into the endoplasmic reticulum where they fold into three dimensional structures with the help of a series of molecular chaperones and folding catalysts. Correctly folded proteins are then transported to the Golgi complex and then delivered to the extracellular environment. However, incorrectly folded proteins are detected by quality control mechanism and sent along another pathway (the unfolded protein response), in which they are ubiquitinated and then degraded in the cytoplasm by the proteasomes [51]. Failure to fold correctly, or to remain correctly folded, gives rise to malfunction in living systems and disease. Proteins with a high propensity to misfold often escape protective mechanisms and form intractable aggregates with themselves, more commonly in the extracellular space. The proteins affected in AD, PD and Prion diseases accumulate in the brain. The ability to form amyloid fibrils seems to be generic; the propensity to do so varies markedly between different amino acid sequences. The relative aggregation rates for a wide range of peptides and proteins correlates with physiochemical features of the molecules such as charge, secondary structure and hydrophobicity [51].
The endoplasmic reticulum is an extensive tubular-reticular network which deals with proteins that are misfolded or aggregated. Normal folding and post-translational modifications are essential for normal cellular functions. If the proteins and peptides are folded, then the unfolded protein response is a mechanism in which cells can handle abnormal proteins. This unfolded protein response is essential to handle the cell’s responses to environmental factors such as injury, immunity and inflammation, and if misfolded proteins are not properly managed, it might lead to disease and cell death [52].
If the unfolded protein response is ineffective it might result in neurodegenerative disorders. There is evidence that overactivity of protein kinase RNA-like ER kinase (PERK, encoded by EIF2AK3) directly contributes to pathological processes that are critical in the reduction of neuronal proteins involved in learning and memory [53].
The unfolded protein response might involve a number of different mechanisms including the inositol requiring enzyme 1A, the PKR-like ER kinase dependent phosphorylation, and the ATF6A which enable the removal of misfolded proteins from the endoplasmic reticulum [52]. Molecular chaperones seem to be important in this process: chaperone triggering factors seem to prevent peptides and proteins from misfolding as they emerge from the endoplasmic reticulum by influencing the hydrophobic residues and to protect them from the cell’s polar interior stopping their misfolding and potential for diseasechaperone function therefore is also important in the unfolded protein response and the mechanisms of neurodegeneration and stochasticity (Figure 3).
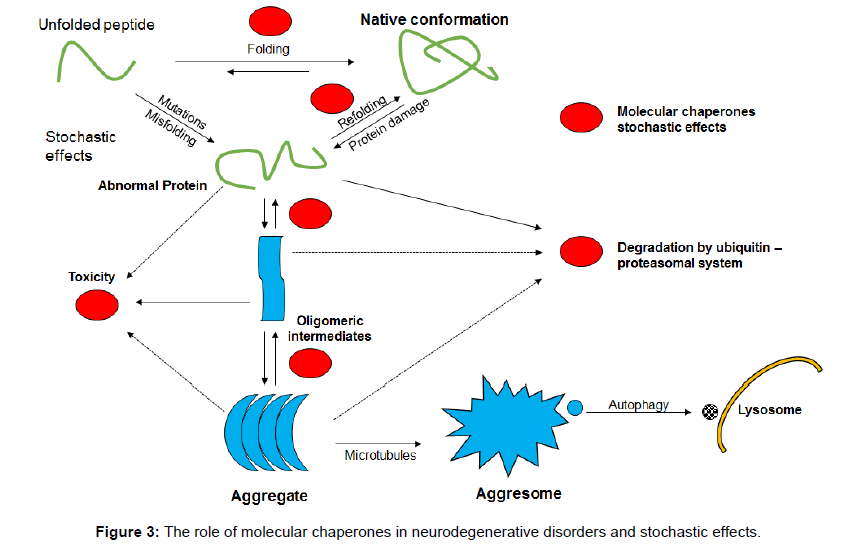
Figure 3: The role of molecular chaperones in neurodegenerative disorders and stochastic effects.
Heat shock proteins and, in particular, heat shock protein 70 (Hsp70 – a molecular chaperone) are up-regulated by different pathological mechanisms and defend the proteome. Hsp70 stabilizes lipid membranes and helps endocytosis, prevents apoptosis, enhances cellular survival and facilitates interaction with the immune system [54]. These membrane and lipid associated functions of Hsp70, if disrupted in pathological states like AD and other neurodegenerative disorders, might prevent autophagy/lysomal dysfunction leading to neuronal death from the aggregation of toxic proteins.
It is important to stress that molecular chaperones help to stop misfolding and to restore proteins to their normal shape. Identification of abnormal proteins by the ubiquitin proteasome system also involves chaperones. A conformational effect of chaperones makes polypeptides and proteins less soluble and unable to be incorporated into the degradation aggresome system leading to disease. Chaperones also disturb signalling pathways that stimulate apoptosis [55-60].
There are many protein variants generated from a limited number of genes. There are several million proteins in the human body generated from about 15,000 genes. How do these proteins arise? The protein variants make up the proteoform- which arise from single genes and represent a unique combination of amino acid sequences with variations. This proteoform variation arises from several mechanisms: alternative splicing, endogenous proteolytic processing and posttranslational modifications to generate the proteoform. There are a number of possible proteoforms from a single coding gene and only one or a few sequence variations that correlate with disease. Proteins are versatile macromolecules with a wide range of functions including catalysis, regulation, communication, mechanical support and movement of transport. The generation of proteoform diversity has major biological significance and represents a significant amplification of biological information from gene to proteoform.
It is postulated the stochastic processes are operative in proteoform generation. Our research seeks to analyse the nature of this stochastic variation in protein sequence variation. Genetically, identical cells exposed to the same environmental conditions show variation in molecular content. This variability is linked to stochasticity in gene expression, which is generally viewed as having detrimental effects on cell function and implications for disease. Stochasticity can, however, be advantageous providing flexibility for cells to adapt in fluctuating environments or respond to sudden stress, such that a mechanism which populates heterogeneity can be established during cellular differentiation and development [61].
In gene expression, there are a small number of molecules involved such as DNA, mRNA and regulatory proteins; gene expression is a stochastic phenomenon. In eukaryotic cells the stochastic effects primarily originate in the regulation of gene activity. Transcription can be initiated by a single transcription factor binding to a specific regulatory site in the target gene. Stochasticity of transcription factor binding and dissociation are then amplified by transcription and translation since target gene activation results in a burst of mRNA molecules and each mRNA copy provides a template for translating numerous protein molecules. Mathematical models have been developed for stochastic processing and this includes ordinary differential equations with a stochastic component of mRNA and protein levels in a single cell, which yield a system of first order partial differential equations for twodimensional probability density functions [62]. Protein production involves a series of stochastic chemical steps. One consequence of this is that the copy number of any given protein varies substantially from one cell to another, even within isogenic populations. Experiments have measured this variation for thousands of different proteins revealing a linear relationship between variants and mean level expression for much of the proteome. This relationship is thought to arise from the random production and degradation of mRNAs, but several lines of evidence suggest that infrequent gene activation events also bear responsibility [63]. Many cellular components and molecules are present in low numbers in a cell and that random births and deaths of individual molecules can cause “noise”. Biochemical events, however, do not necessarily occur in single steps of individual molecules. Some processes are greatly randomised when synthesis or degradation occurs in large bursts of many molecules during a short time interval. Each birth or death of a macromolecule could involve several small steps creating a memory between individual events. A generalised theory for stochastic gene expression has been proposed, following the variants in protein abundance in terms of the randomness of the individual gene expression events. It has been shown that common types of molecular mechanisms can produce gestation and senescence periods that induce noise without requiring higher abundance, shorter lifetimes or any concentration dependent control loops [64]. Methodologically, single cell experiments cannot distinguish qualitatively different stochastic principles, although this in turn makes such methods better suited for identifying which component introduced fluctuations- probably dynamic measurements with single molecule resolution are required. Experimental techniques have followed gene expression in single cells over time and reveal stochastic bursts of both mRNA and protein synthesis in many different types of organisms. Stochasticity has been shown to be exploited by bacteria and viruses to decide between different behaviours. In fluctuating environments, cells that respond stochastically can out-compete those that sense environmental changes, and stochasticity may even have contributed to chromosomal gene order [65]. It is proposed that neurons and other cells in the brain utilise these stochastic principles to generate cellular order in preparation for stress. However, stochasticity is important for cellular survival and occasionally the protein sequence, which is randomly produced and has sequence variation, leads to disease by favouring misfolding and aggregation. More recently human proteins have been mapped across the cell. The location of around 12,000 proteins among 13 sub-cell localisations have identified where the biomolecules reside. In the examination of 22 human cell lines, antibody-based fluorescence microscopy identified that many proteins locate in multiple places around the cell from mitochondria to other entities from cellular junctions, vesicles, nuclear membranes, microtubules and a newer organelle known as the aggresome, where a cell’s misfolded proteins are collected for processing before degradation [66]. It is postulated that the stochastically generated abnormal proteins become misfolded and are resistant to the aggresome. This resistance coupled with biophysical and other variables, including over-production, leads to disease.
Discussion
Prion diseases occur by misfolding of prion proteins, which spread by a seeding process where one misfolded aggregate can seed the misfolding in other normally folded molecules by a mechanism known as seeded polymerisation [67]. The seeded amplification results in increased levels of the misfolded protein and spread to adjacent brain regions. In addition, extracts from these brains can transmit prion protein to new individuals experimentally, iatrogenicly or by natural routes. The realisation that seeded polymerisation is a similar process not only for infectious prion disease, but some in the noninfectious neurological diseases has led to the prion-like effects of neurodegenerative disorders [68]. Recently, it has been shown that AD amyloid proteins may spread from surgical instruments, where it was observed that children having neurosurgical procedures developed cerebral amyloid angiopathy decades later [69]. Prion diseases are slowly progressive fatal brain diseases in which vacuoles develop in the grey matter with prominent gliosis involving microglia and deposition of aggregated protease-resistant PrPSC or PrPRES isoforms derived from host-encoded normal prion protein [PrPC or PrPSEN]. These diseases occur spontaneously in humans and ruminants and can be transmitted. Within a given animal species, multiple strains of prion infectivity have been identified based primarily on different patterns of regional brain pathology as the clinical end point. The molecular explanation for the maintenance of diverse strain phenotypes with only one type of PrP protein sequence is not certain. However, the secondary structure of the PrPRES aggregates is known to differ among certain strains and such structures appear to be maintained during template replication of proteins using a single primary PrP protein sequence [70].
Most studies of prion strains have focused on strain-specific differences in the regional patterns of prion-induced vacuolar neuropathology and/or PrPSC deposition, but some observers have described strain differences in association with PrPSC in particular brain cell types. For example, sheep infected naturally or experimentally with sheep or mouse scrapie were found to have strain-specific patterns of PrPSC accumulation within neurons or glia. In hamster experiments, accumulation of PrPSC in neuronal soma at the clinical end point varied among 8 scrapie strains and appeared to correlate with shorter incubation periods. In other studies using mice, morphological patterns of PrPSC deposition were shown to differ among different scrapie strains; for example, ME7 was primarily neuronal, and 79A was both neuronal and astroglial. Although certain patterns of cell association were clear in these experiments, the extensive spread and deposition of PrPSC at the clinical end point might obscure the initial specificity of PrPSC for certain brain structures or cells [67].
The Microtubule Associated Protein Tau (MAPT) belongs to a family of homologous proteins, including MAP2 and MAP4, with three or four basic microtubule binding domains in their carboxy terminal regions. The amino terminus may also interact with microtubules, but precise functional interactions are not well understood. The three members of the MAPT/MAP2/MAP4 family expressed as multiple splice variants, some of which contain different numbers of microtubule binding domains. MAPT and MAP2 are expressed mainly in neurons where they show a characteristic subcellular compartmentalisation, with MAP2 being somatodendritic, MAPT predominantly present in the axon and MAP4 a major non-neuronal microtubule-associated protein. MAPT is what is referred to as a intrinsically disordered protein which can adopt dynamic conformations. Intrinsically disordered proteins account for a substantial proportion of the proteome and many of them are promiscuous binders that undergo a partial transition from an ordered state in which they interact stably with various partners and frequently function as molecular hubs in protein interaction networks [71-76].
Primary and posttranslational modifications of MAPT can compromise its physiological role in microtubule assembly and in mediating other cellular functions [76-79]. Such considerations might contribute to the aggregate formation in central neurons that are pathognomonic for AD and other “tauopathies” and could create MAPT species with toxic properties [80]. The regulation of MAPT expression and epigenetic contributions remain to be explored and alternative splicing patterns depend on species, tissue and condition [81,82]. The dynamic internal and external interactions of MAPT are influenced by primary sequence variation, post-translational modifications and polarised charge distribution that determine its site-specific properties responsible for physiological function and neuropathogenic effects [71,76,81-84]. Mechanistic studies will unravel the complexity inherent in tau aggregation leading to AD and the tauopathies.
AD can occur as a result of genetic mutation or, more commonly, occurs as a sporadic event. Young-onset disease is probably a result of a stochastic change in the Aβ or microtubule-associated tau protein leading to misfolding and aggregation. Either is possible in that APP mutations can result in AD; tau follows, so, alternatively, tau can aggregate and might affect the function of Aβ leading to pathogenic fibrils [18]. Young-onset and late-onset AD might begin in specific regions of the brain and the studies of Qiang et al. support that there might be species of Aβ molecules that determines the disease aggressiveness and site-specific origin such as posterior cortical atrophy [5,16]. This concept is similar to that of the Prion diseases.
It is also speculated that in disorders like PD, the second most common sporadic neurodegenerative disorder, α-synuclein, a peptide that can also undergo oligomerization and fibrillar formation, can start at particular parts in the brain; e.g., substantia nigra, leading to traditional PD or in the cerebral cortex leading to dementia with Lewy body disease, both of which are part of a spectrum. Similar considerations might explain why some patients present with unilateral or lower limb disease. This approach probably accounts for the deposition of tau in other tauopathies such as FTD, including Progressive Supranuclear Palsy (PSP). That is, the anatomical site for the stochastically impaired protein leading to misfolding and, in a certain microenvironmental milieu, neurodegeneration. Similarly, in FTD another entity related to tau, TDP-43 or C9orf72, the initial molecular step might occur, say in the right temporal lobe in FTD, leading to the right temporal variant, in primary progressive aphasia within the left temporal lobe linguistic variants such as primary progressive non-fluent aphasia or semantic dementia; it can also affect the frontal lobes leading to the behavioural variant of FTD [85-88].
In conclusion, stochasticity is fundamental to the development of sporadic neurodegenerative disorders. An abnormal peptide sequence or other molecular variation, results in abnormal folding in a particular part of the brain (that is, anatomical specificity) as a result of the peptides’ biophysical features and intracellular milieu. The peptide folds in a certain way and, due to microenvironmental influences, and the biophysical properties of the misfolded protein, creates nuclear effects such that the misfolding peptide causes overproduction of the aberrant sequence with its unique physicochemical characteristics, that then sets a chain reaction generating aggregation of the aberrant misfolded protein, which bypasses the cell’s normal mechanisms, such as the proteasome/aggresome system. The aggregated misfolded protein is excreted into the extracellular environment, taken up by surrounding cells, resulting in the progression of neurodegeneration. This process may be stimulated by head injury, stress, APOE and infection including Herpes virus or, as recently discovered, the bacteria Porphyromonas gingivalis, one of the principle causes of chronic periodontitis [89-91]. There may be contribution from long noncoding RNAs (IncRNAs) in this process [92].
This process can be summarised in a matrix equation (Figure 4). In the case of young-onset AD and other early-onset neurodegenerative disorders, that are not genetic, the nature of the sequence change and misfolding in certain microenvironmental and biophysical circumstances, lack of normal degradation and overproduction leads to an aggressive neurodegenerative disease. In the old onset group, with time the cellular mechanisms such as aggresomal and chaperone functions become less capable, leading to the neurodegenerative process.
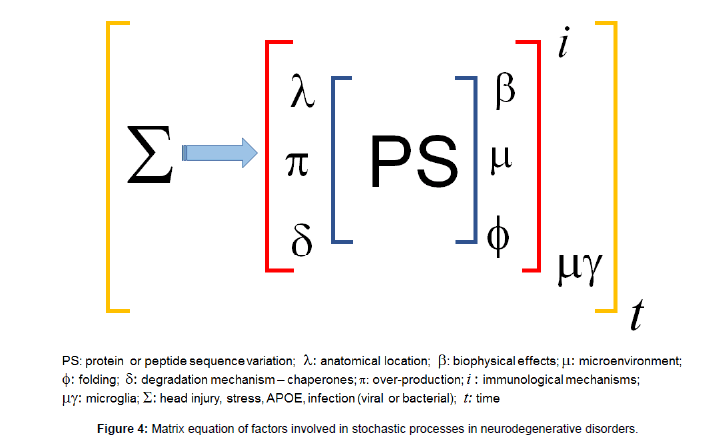
Figure 4: Matrix equation of factors involved in stochastic processes in neurodegenerative disorders.
Our conclusions are supported by discoveries that age and neurodegeneration increase mutations in single human neurons [93- 95]. The work of Gonitel et al. supports the notion that mutations can occur in terminally differentiated neurons and DNA polymorphism may be produced in adult neurons [96]. Furthermore, there is evidence that genome instability may occur in AD, along with DNA repair deficiency that might result in disease [97]. Support from this concept comes from the work of Lee et al. where it was shown that somatic APP gene combination occurs in AD where neurons from individual patients with sporadic AD showed increased genomic cDNAs, with cells having thousands of genomic cDNA variants [98]. These cDNAs lack introns and range from full length cDNA sequences, brain specific RNA splice variants and small forms that contain intron-exon junctions, deletions, exertions and single nucleotide variations. These authors used in situ hybridisation to show that these cDNAs in single neurons were not present in non-neuronal cells or cells from individuals without AD. Retroinsertion of RNA, transcription, breaks in DNA and reversetranscriptase activity were proposed as mechanisms for the origins of genomic cDNA variants [98].
Our considerations of the importance of stochastic processes in gene and protein expression in the brain support the concept that stochastic principles are probably important in neuronal functions in general [99]. Our findings hint at the importance of these processes in brain evolution [100-109]. Our conclusions are supported by recent findings that age and neurodegeneration increase mutations in single human neurons [93].
Conclusion
The considerations presented in this paper suggest avenues for further research and the possibilities of new treatments for neurodegenerative disorders. Experimental studies in the future, using measurements in single cells, will help to answer some of the questions raised in this work.
Funding
This work was supported by Neurodegenerative Disorders Research Pty Ltd.
Acknowledgement
Mariella Panegyres for assistance with the figures.
References
- Xu J, Zhang Y, Qiu C, Cheng F (2017) Global and regional economic costs of dementia: A systematic review. Lancet 390: 4S1-S106.
- Atkins E, Bulsara MK, Panegyres PK (2012) Cerebrovascular risk factors in early-onset dementia. JNNP 83: 666-667.
- Atkins ER, Bulsara MK, Panegyres PK (2012) The natural history of early-onset dementia: The Artemis Project. BMJ Open 2: e001764.
- Panegyres PK, Goh JGS, McCarthy M, Campbell A (2017) The Nature and Natural History of Posterior Cortical Atrophy Syndrome. Alzheimer Dis Assoc Disord 31: 295-306.
- Jarmolowicz AI, Chen HY, Panegyres PK (2015) The Patterns of Inheritance in Early Onset Dementia: Alzheimer’s Disease and Frontotemporal Dementia. Am J Alzheimers Dis Other Demen 30: 299-306.
- Cacace R, Sleegers K, Van Broeckhoven C (2016) Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dem 12: 733-748.
- Perrone F, Cacace R, Van Mossevelde S, Van den Bossche T, De Deyn PP, et al. (2018) Genetic screening in early-onset dementia patients with unclear phenotype: Relevance for clinical diagnosis. Neurobiol Aging 69: 292.e7-292.e14.
- Alonso Vilatela ME, López-López M, Yescas-Gómez P (2012) Genetics of Alzheimer's disease. Arch Med Resh 43: 622-631.
- Chai C, Lim K-L (2013) Genetic insights into sporadic Parkinson’s disease pathogenesis. Curr Gen 14: 486-501.
- Safar JG (2012) Molecular pathogenesis of sporadic prion diseases in man. Prion 6: 108-115.
- Shaw PJ (2005) Molecular and cellular pathways of neurodegeneration in motor neurone disease. JNNP 76: 1046-1057.
- Capozzo R, Sassi C, Hammer MB, Arcuti S, Zecca C, et al. (2017) Clinical and genetic analyses of familial and sporadic frontotemporal dementia patients in Southern Italy. Alzheimers Dem 13: 858-869.
- Rosso SM, Landweer EJ, Houterman M, Donker Kaat L, van Duijn CM, et al. (2003) Medical and environmental risk factors for sporadic frontotemporal dementia: a retrospective case-control study. JNNP 74: 1574-1576.
- Qiang W, Yau W-M, Lu J-X, Collinge J, Tycko R (2017) Structural variation in amyloid-ß fibrils from Alzheimer’s disease clinical subtypes. Nature 541: 217-221.
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: 239-59.
- Goedert M (2015) Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aß, tau, and a-synuclein. Science 349: 1255555.
- Xiao B, Cui L-Q, Chen T-M, Lianb B (2015) Stochastic effects in adaptive reconstruction of body damage: implied the creativity of natural selection. J Cell Mol Med 19: 2521-2529.
- Elowitz MB, Levine AJ, Siggia ED, Swain PS (2002) Stochastic gene expression in a single cell. Science 297: 1183-1186.
- Churchill GA (1989) Stochastic models for heterogeneous DNA sequences. Bull Math Biol 51: 79-94.
- Bogarad LD, Deem MW (1999) A hierarchical approach to protein molecular evolution. Proc Natl Acad Sci USA 96: 2591-2595.
- Nei M (2005) Selectionism and neutralism in molecular evolution. Mol Biol Evol 22: 2318-2342.
- Bauer AL, Jackson TL, Jiang Y, Rohlf T (2010) Receptor cross-talk in angiogenesis: Mapping environmental cues to cell phenotype using a stochastic, Boolean signaling network model. J Theoretical Biol 264: 838-846.
- Moghal A, Mohler K, Ibba M (2014) Mistranslation of the genetic code. FEBS Lett 588: 4305-4310.
- Bullwinkle TJ, Reynolds NM, Raina M, Moghal A, Matsa E, et al. (2014) Oxidation of cellular amino acid pools leads to cytotoxic mistranslation of the genetic code. ELIFE 3: e02501.
- Bezerra AR, Simões J, Lee W, Rung J, Weil T, et al. (2013) Reversion of a fungal genetic code alteration links proteome instability with genomic and phenotypic diversification. Proc Natl Acad Sci USA 110: 11079-11084
- Nam K, Mugal C, Nabholz B, Schielzeth H, Wolf JBW, et al. (2010) Molecular evolution of genes in avian genomes. Genome Biol 11: R68.
- Pechmann S, Frydman J (2014) Interplay between Chaperones and Protein Disorder Promotes the Evolution of Protein Networks. PLOS Comput Biol 10: e1003674.
- Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, et al. (2015) A human interactome in three quantitative dimension organized by stochiometries and abundances. Cell 163: 712-723.
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, et al (2017) Architecture of the human interactome defines protein communities and disease networks. Nature 545: 505-509.
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, et al. (2015) The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell 162: 425-440.
- Sharan R, Suthram S, Kelley RM, Kuhn T, McCuine S, et al. (2005) Conserved patterns of protein interaction in multiple species. Proc Natl Acad Sci USA 102: 1974-1979.
- Kutschera U, Niklas KJ (2004) The modern theory of biological evolution: an expanded synthesis. Naturwissenschaften 91: 255-276.
- McAdams HH, Arkin A (1997) Stochastic mechanisms in gene expression. Proc Natl Acad Sci USA 94: 814-819.
- Martin GM (2012) Stochastic modulations of the pace and patterns of ageing: impacts on quasi-stochastic distributions of multiple geriatric pathologies. Mech Ageing Dev 133: 107-111.
- Kosik KS (2006) The neuronal microRNA system. Nat Rev Neurosci 7: 911-920.
- Rahman M, Previs SF, Kasumov T, Sadygov RG (2016) Gaussian process modelling of protein turnover. J Proteome Res 15: 2115-2122.
- Makrides SC (1983) Protein synthesis and degradation during aging and senescence. Biological Rev 58.
- Rodriguez F, Oliver JL, Marin A, Medina JR (1990) The general stochastic model of nucleotide substitution. J Theor Biol 142: 485-501.
- Vallender EJ (2008) Exploring the origins of the human brain through molecular evolution. Brain Behav Evol 72: 168-177.
- Bae T, Tomasini L, Mariani J, Zhou B, Roychowdhury T, et al. (2018) Different mutational rates and mechanisms in human cells at pre-gastrulation and neurogenesis. Science 359: 550-555.
- Lodato MA, Woodworth MB, Lee S, Evrony GD, Mehta BK, et al. (2015) Somatic mutation in single human neurons tracks developmental and transcriptional history. Science 350: 94-98.
- Lee JH (2018) Tracing single-cell histories: Origins of mutations in single cells during human brain development and aging are revealed. Science 359: 521-522.
- Park JS, Lee J, Jung ES, Kim M-H, Kim IB, et al. (2019) Brain somatic mutations observed in Alzheimer’s disease associated with aging and dysregulation of tau phosphorylation. Nat Commun 20: 3090.
- Caniparoli L, Lombardo P (2014) Nonequilibrium stochastic model for tRNA binding time statistics. Phys Rev 89: 012712.
- Reeves JH (1992) Heterogeneity in the substitution process of amino acid sites of proteins coded for by mitochondrial DNA. J Mol Evol 35: 17-31.
- Gal-Ben-Ari S, Kenney JW, Ounalla-Saad H, Taha E, David O, et al. (2012) Consolidation and translation regulation. Learn Mem 19: 410-422.
- Grootjans J, Kaser A, Kaufman RJ, Blumberg RS (2016) The unfolded protein response in immunity and inflammation. Nat Rev Immunol 16: 469-484.
- Smith HL, Mallucci GR (2016) The unfolded protein response: Mechanisms and therapy of neurodegeneration. Brain 139: 2113-2121.
- Balogi Z, Multhoff G, Jensen TK, Lloyd-Evans E, Yamashima T, et al. (2019) Hsp70 interactions with membrane lipids regulate cellular functions in health and disease. Progress in Lipid Res 74: 18-30.
- Merlin AB, Sherman MY (2005) Role of molecular chaperones in neurodegenerative disorders. Int J Hyperthermia 21: 403-419.
- Lindberg I, Shorter J, Wiseman RL, Chiti F, Dickey CA, et al. (2015) Chaperones in neurodegeneration. J Neurosci 34: 13853-13859.
- Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, et al. (2017) Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener 6: 6.
- Raj K, Chanu SI, Sarkar S (2015) Protein misfolding and aggregation in neurodegenerative disorders: Focus on chaperone-mediated protein folding machinery. Int J Neurol Res 2: 72-78.
- Guerriero CJ, Brodsky JL (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol Rev 92: 537-576.
- Wang M, Kaufman RJ (2016) Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 529: 326-335.
- Kaern M, Elston TC, Blake WJ, Collins JJ (2005) Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet 6: 451-464.
- Lipniacki T, Paszek P, Marciniak-Czochra A, Brasier AR, Kimmel M (2006) Transcriptional stochasticity in gene expression. J Theoretical Biol 238: 348-367.
- Kaufmann BB, van Oudenaarden A (2007) Stochastic gene expression: from single molecules to the proteome. Curr Opin Genet Dev 17: 107-112.
- Pedraza JM & Paulsson J (2008) Effects of molecular memory and bursting on fluctuations in gene expression. Science 319: 339-343
- Shahrezaei V, Swain PS (2008) The stochastic nature of biochemical networks. Curr Opin Biotechnol 19: 369-374.
- Thul PJ, Ã…kesson L, Wiking M, Mahdessian D, Geladaki A, et al. (2017) A subcellular map of the human proteome. Science 356: pii: eaal3321.
- Carroll JA, Striebel JF, Rangel A, Woods T, PhillipsK, et al. (2016) Prion strain differences in accumulation of PrPSc on neurons and glia are associated with similar expression profiles of neuroinflammatory genes: Comparison of three prion strains. PLOS Pathogens 12: e1005551.
- Kraus A, Groveman BR, Caughey B (2013) Prions and the potential transmissibility of protein misfolding diseases. Annu Rev Microbiol 67: 543-564.
- Jaunmuktane Z, Quaegebeur A, Taipa R, Viana-Baptista M, Barbosa R, et al. (2018) Evidence of amyloid- ß cerebral amyloid angiopathy transmission through neurosurgery. Acta Neuropathol 135: 671-679.
- Caughey B, Raymond GJ, Bessen RA (1998) Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem 273: 32230-32235.
- Sündermann F, Fernandez M-P, Morgan RO (2016) An evolutionary roadmap to the microtubule-associated protein MAP Tau. BMC Genomics 17: 264.
- Fichou Y, Heyden M, Zaccai G, Weik, Tobias DJ (2015) Molecular dynamics simulations of a powder model of the intrinsically disordered protein tau. J Phys Chem 119: 12580-12589.
- Li X-H, Culver JA, Rhoades E (2015) Tau binds to multiple tubulin dimers with helical structure. J Am Chem Soc 137: 9218-9221
- Wright PE & Dyson HJ (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nature 16: 18-29.
- Kadavath H, Jaremko M, Jaremko L, Biernat J, Mandelkow E, et al. (2015) Folding of the tau protein on microtubules. Angew Chem Int Ed 54: 10347-10351.
- Avila J, Lucas JJ, Perez M, Hernandez F (2004) Role of tau protein in both physiological and pathological conditions. Physiol Rev 84: 361-384.
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, et al. (2010) Dendritic function of tau mediates amyloid- ß  toxicity in Alzheimer’s mouse models. Cell 142: 387-397.
- Morris M, Maeda S, Vossel K, Mucke L (2011) The many faces of tau. Neuron 70: 410-426.
- Fontaine SN, Sabbagh JJ, Baker J, Martinez-Licha CR, Darling A, et al. (2015) Cellular factors modulating the mechanism of tau protein aggregation. Cell Mol Life Sci 72: 1863-1879.
- Shahani N, Subramaniam S, Wolf T, Tackenberg C, Brandt R (2006) Tau aggregation and progressive neuronal degeneration in the absence of changes in spine density and morphology after targeted expression of Alzheimer’s disease-relevant tau constructs in organotypic hippocampal slices. Neurobiol Dis 26: 6103-6114.
- Andreadis A (2012) Tau splicing and the intricacies of dementia. J Cell Physiol 227: 1220-1225.
- Caillet-Boudin M-L, Buee L, Sergeant N, Lefebvre B (2015) Regulation of human MAPT gene expression. Mol Neurodegener 10: 28.
- Mandelkow E-M, Mandelkow E (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2: a006247.
- Ramachandran G, Udgaonkar JB (2013) Mechanistic studies unravel the complexity inherent in tau aggregation leading to Alzheimer’s disease and the tauopathies. Biochem 52: 4107-4126.
- Verstraeten A, Theuns J, Van Broeckhoven C (2015) Progress in unravelling the genetic etiology of Parkinson disease in a genomic era. Trends in Genetics 31: 140-149.
- Tinh J, Farrer M (2013) Advances in the genetics of Parkinson disease. Nat Rev Neurol 9: 445-454.
- Sieben A, Van Langenhove T, Engelborghs S, Martin JJ, Boon P, et al (2012) The genetics and neuropathology of frontotemporal lobar degeneration. Acta Neuropathol 124: 353-372.
- Pottier C, Ravenscroft TA, Sanchez-Contreras M, Rademakers R (2016) Genetics of FTLD: Overview and what else we can expect from genetic studies. J Neurochem 138 S1: 32-53.
- Readhead B, Haure-Mirande J-V, Funk CC, Ehrlich ME, Gandy S, et al. (2018) Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99: 64-82.
- Itzhaki RF (2018) Corroboration of a major role for Herpes simplex virus type 1 in Alzheimer’s disease. Frontiers of Aging Neurosci 10: 324.
- Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, et al. (2019) Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv 5: eaau3333.
- Wan P, Su W, Zhuo Y (2017) The role of long noncoding RNAs in neurodegenerative diseases. Mol Neurobiol 54: 2012-2021.
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, et al. (2018) Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359: 555-559.
- McConnell MJ, Moran JV, Abyzov A, Akbarian S, Bae T, et al. (2017) Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science 356: eaal1641.
- Hazen JL, Faust GG, Rodriguez AR, Kupriyanov S, Hall IM, et al. (2016) The complete genome sequences, unique mutational spectra, and developmental potency of adult neurons revealed by cloning. Neuron 89: 1223-1236.
- Gonitel R, Moffitt H, Sathasivam K, Woodman B, Detloff PJ, et al. (2008) DNA instability in postmitotic neurons. PNAS 105: 3467-3472.
- Hou Y, Song H, Croteau DL, Akbari M, Bohr VA (2016) Genome instability in Alzheimer’s disease. Mech Ageing Dev 161: 83-94.
- Lee MH, Siddoway B, Kaeser GE, Segota I, Rivera R, et al. (2018) Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature 563: 639-645.
- Fay JC, Wu C-I (2003) Sequence divergence, functional constraint, and selection in protein evolution. Annu Rev Genomics Hum Genet 4: 213-235.
- Acar M, Mettetal JT, van Oudenaarden A (2008) Stochastic switching as a survival strategy in fluctuating environments. Nat Genet 40: 471-475.
- Loftfield RB, Vanderjagt D (1972) The frequency of errors in protein biosynthesis. Biochem J 128: 1353-1356.
- Guo HH, Choe J, Loeb LA (2004) Protein tolerance to random amino acid change. PNAS 101: 9205-9210.
- Nei M, Suzuki Y, Nozawa M (2010) The natural theory of molecular evolution in the genomic era. Ann Rev Genom Hum Genet 11: 265-289.
- Akashi H, Osada N, Ohta T (2012) Weak selection and protein evolution. Genetics 192: 15-31.
- Milias-Argeitis A, Engblom S, Bauer P, Khammash M (2015) Stochastic focusing coupled with negative feedback enables robust regulation in biochemical reaction networks. J Royal Soc Interface 12: 20150831.
- Yu J-F, Cao Z, Yang Y, Wang C-L, Su Z-D, et al (2016) Natural protein sequences are more intrinsically disordered than random sequences. Cell Mol Life Sci 73: 2949-2957.
- Raj A, van Oudenaarden A (2008) Stochastic gene expression and its consequences. Cell 135: 216-226.
- Drummond DA, Wilke CO (2009) The evolutionary consequences of erroneous protein synthesis. Nat Rev Genet 10: 715-724.
Citation: Panegyres PK (2019) Stochastic Considerations into the Origins of Sporadic Adult Onset Neurodegenerative Disorders. J Alzheimers Dis Parkinsonism 9: 473. DOI: 10.4172/2161-0460.1000473
Copyright: © 2019 Panegyres PK, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 5340
- [From(publication date): 0-2019 - Dec 19, 2025]
- Breakdown by view type
- HTML page views: 4351
- PDF downloads: 989