Excessive Intake Salt Contribution to Cognitive Impairment in Mice
Received: 05-Mar-2015 / Accepted Date: 15-Apr-2015 / Published Date: 22-Apr-2015 DOI: 10.4172/2329-9053.1000124
Abstract
Dementia mice model was prepared by D-gal, Morris water maze was used for behavior research; c(WB) method was used for the pre-dementia protein expression research. Results showed that excessive intake of salt significantly decreased the memory ability and significantly prolonged the escape time and distance to get the platform; WB results showed that expressions of beta amyloid (Aβ) and β-site APP cleaving enzyme 1 (BACE1) significantly increased and the NEP expression significantly decreased under the condition of excessive salt intake. Results suggest that excessive salt intake can regulate the protein expressions of Aβ, BACE1 and neprilysin to enhance D-gal-induced dementia.
Keywords: Edible salt; D-gal; Aging; Dementia
1650Introduction
Alzheimer's disease (AD) is a syndrome characterized by declining ability to learn and memory function. A variety of clinical methods are used for detection and diagnosis, but these methods are inadequate for the detection of early stage dementia. Recent research has shown that the expressions of beta amyloid (Aβ) deposition-associated molecules could be used as early stage biomarkers [1]. These include increased expression of the Aβ and β-site APP cleaving enzyme 1 (BACE1) and decreased expression of neprilysin (NEP). All these biomarkers occurred much earlier than obvious memory injury in mice.
D-galactose (D-gal) is a dialdehyde sugar that can be metabolized at the body's normal concentration as a normal nutrient; however, the oxidase or galacturonic dehydrogenase can be generated at high levels of D-gal, and may be further metabolized to xylulose while generating superoxide anions and oxygen-derived free radicals. Excess oxygen free radicals can cause oxidative stress. Advanced glycation end products (AGEs) could also be formed by D-galactose readily reacting with the free amines of amino acids in proteins and peptides. Evidence shows that AGEs can significantly cause reactive oxygen species (ROS) accumulation, especially superoxide radicals and hydrogen peroxide. An animal model of oxidative stress has used mice injected with D-gal [2,3]. Recently, oxidative stress mediated DNA damage and alterations in synaptic structure and memory maintenance as well as in learning abilities were also demonstrated in mouse models.
Evidence has shown that a number of health conditions are caused by, or exacerbated by, a high edible salt (ES) diet. Although the strongest evidence is for effects on blood pressure, stroke and heart disease, there is also a wide body of evidence indicating a link between ES consumption and other conditions [4,5]. ES is also thought to exacerbate the symptoms of AD. In this study, we investigated this phenomenon by using the model of AD induced by D-gal in mice.
Materials And Methods
Animal experiment
The experiment was carried out on adult male KunMing mice (18–22 g). The mice were housed in a quiet, temperature- and humidity-controlled room (22 ± 2°C and 60 ± 5%, respectively) with a 12-h light/dark cycle with food and water continuously available. All experiments were performed at the same time of day during the period of light. All procedures in the present study were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adapted by the National Institutes of Health (Washington, DC, USA, 1996). Local ethics committee approval was also obtained (Ethic committe number: 10/2013). All efforts were made to minimize animal suffering and to reduce the number of animals used. Sixty animals were purchased from the Animal Center of Jilin University and adapted to the setting for 7 days. The animals were then randomly divided into four treatment groups (control (Con), D-gal (D-G), D-gal plus low ES (D-GL), and D-gal plus high ES (D-GH). D-gal (300 mg/ kg) was injected subcutaneously into the mice daily for 8 weeks, and low ES (the equivalent of people: 6 g/70 kg body weight) and high ES (the equivalent of people: 18 g/70 kg body weight) were gavaged 10 h prior to D-gal injection. All Con group animals were given saline intraperitoneally. (To determine memory impairment caused by D-gal plus ES, we tested whether ES can weaken spatial memory function by using the Morris water maze. The animals were tested 10 times (2 times/5 days) in the maze. The mice were sacrificed on the last day of treatment, and sera and major organs were collected immediately (Figure 1).
Figure 1: Experimental scheme.
Chemicals and reagents
The NuPAGE Bis-Tris Electrophoresis System (precast polyacrylamide mini gel) was purchased from Bio-Rad (USA). COX-2 antibody was purchased from Thermo scientific (USA). PARP antibodies and horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG secondary anti-bodies, polyvinyl difluoride (PVDF) membranes, the BSA protein assay kit, Western Blot chemiluminescence reagent, the superoxide dismutase activity assay kit, the glutathione peroxidase assay kit, and the DNA Fragmentation Assay Kit were all purchased from JianCheng (Nanjing, China).
Morris water maze test
The water maze test is a widely accepted method for testing memory, and we utilized this to determine memory impairment. Maze testing was performed in accordance with the TaiMeng program and equipment (ChengDu, China). A quiet environment, consistent lighting, constant water temperature and a fixed spatial frame were maintained throughout the period of the experiment [6].
Brain tissue collection and preservation
After the behavioral test, the brains were collected in a consistent manner, frozen at -20°C and separated into cortical and hippocampal regions. All the brain sections were stored at -80°C and then subjected to biological assay.
Measurement of GSH-Px, SOD and MDA levels
Glutathione peroxidase (GSH-Px), superoxide dismutase (SOD) and malondialdehyde (MDA) levels in the serum were determined using ELISA kits (Jiancheng, Nanjing, China) as described in the manufacturer’s manual. Briefly, 100 ul of the sample was pipetted into the primary coated-plate and incubated overnight at 4°C. After each well was washed with washing buffer, 100 ul of secondary antibody solution was added and the mixture was incubated for 1 h at 4°C in the dark. After rinsing, chromogen was added and the mixture was incubated for 30 min at room temperature in the dark. After addition of the stop solution, the resulting color was assayed at 450 nm using a microplate absorbance reader (Sunrise™, TECAN, Switzerland).
Western Blot analysis: The cells were lysed on ice with 200 mL of lysis buffer (50 mM Tris-HCl, pH 7.5, 0.5 M NaCl, 5 mM MgCl2, 0.5% Nonidet P 40, 1 mM phenyl-methylsulfonyl fluoride for, 1 mg/ mL pepstatin, and 50 mg/ml leupeptin) and centrifuged at 13,000 g at 4°C for 20 min. The protein concentrations in the supernatants were quantified using a Nanjing, Jiancheng Protein Assay Kit. Electrophoresis was performed on a NuPAGE Bis-Tris electrophoresis system using 50 mg of reduced protein extract per lane. Resolved proteins were then transferred to polyvinyl difluoride (PVDF) membranes. Filters were blocked with 5% nonfat milk overnight and probed with the appropriate dilution of primary antibodies for 1 h at room temperature. Membranes were washed three times with 0.1% Tween 20 and incubated with HRP-conjugated secondary antibody for 1 h at room temperature. All proteins were detected using Western Lightning Chemiluminescence Reagent Plus and quantified using a densitometer.
Statistical analysis
Data are shown as mean and standard deviation. Statistical differences were analyzed using Student’s t-test for normally distributed values and nonparametric Mann-Whitney t-tests for values in nonnormal distributions. Values of P
Results And Discussion
The D-gal, D-gal plus low ES, and D-gal plus high ES-treated mice arrived at the platform more slowly than did controls, and both D-gal plus low salt and D-gal plus high salt treated groups significantly increased the effects of D-gal on escape latencies (Tables 1 and 2). All of the mice exhibited shorter and shorter escape latency and escape distance by the end of the training trial, as the escape latency to the platform at the end of training was about 5.0 ± 1.07 s and the escape distance was 120.4 ± 22.5 cm after 10 trials in the control group. D-galinduced mice exhibited escape latencies of 14.05 ± 1.78 s and escape distance of 344.6 ± 44.8 cm and. Low ES plus D-gal-induced mice exhibited escape latencies of 27.53 ± 2.79 s, and escape distance of 664.2 ± 51.4 cm; high ES plus D-gal-induced mice exhibited escape latencies of 34.11 ± 3.67 s and escape distance of 854.6 ± 54.3 cm.
| E-T | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 |
|---|---|---|---|---|---|
| A | 22.32 ± 2.34 | 21.15 ± 2.25 | 12.45 ± 2.22 | 7.0 ± 1.23 | 5.0 ± 1.07 |
| B | 35.66 ± 4.68* | 29.53 ± 3.49* | 23.27 ± 2.97* | 19.18 ± 2.22* | 14.05 ± 1.78* |
| C | 42.87 ± 5.42*# | 38.67 ± 4.99*# | 34.56 ± 4.14*# | 30.34 ± 3.67*# | 27.53 ± 2.79*# |
| D | 49.55 ± 6.87*# | 46.34 ± 6.11*# | 42.56 ± 5.33*# | 38.21 ± 4.25*# | 34.11 ± 3.67*# |
Table 1: Escape latency of every group and every day (s).
| E-T | Day 1 | Day 2 | Day 3 | Day 4 | Day 5 |
|---|---|---|---|---|---|
| A | 510.2 ±40.2 | 488.4 ±56.8 | 282.5 ±41.2 | 172.4 ±32.3 | 120.4 ±22.5 |
| B | 843.5 ±89.5* | 702.3 ±66.7* | 552.4 ±65.4* | 464.9 ±46.2* | 344.6 ±44.8* |
| C | 1002.6 ±94.3*# | 928.8 ±77.4*# | 843.2 ±75.5*# | 704.8 ±69.4*# | 664.2 ±51.4*# |
| D | 1225.4 ±119.6*# | 1156.3 ±74.4*# | 995.4 ±69.8*# | 955.4 ±65.5*# | 854.6 ±54.3*# |
Table 2: Escape distance of every group and every day (cm).
As shown in Figure 2, the GSH-Px levels of the D-Gal, D-GL and D-GH groups were significantly decreased compared to the Con group, and the D-GL and D-GH groups were significantly decreased compared to the D-Gal group. The SOD levels of the D-Gal, D-GL and D-GH groups were significantly decreased compared to the Con group and the D-GH group was significantly decreased compared to the D-Gal group. MDA levels of the D-Gal, D-GL and D-GH groups were significantly increased compared to the Con group, and the D-GH group was significantly increased compared to the D-Gal group.
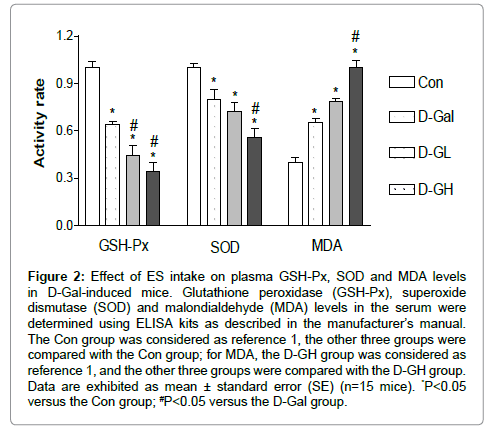
Figure 2: Effect of ES intake on plasma GSH-Px, SOD and MDA levels in D-Gal-induced mice. Glutathione peroxidase (GSH-Px), superoxide dismutase (SOD) and malondialdehyde (MDA) levels in the serum were determined using ELISA kits as described in the manufacturer’s manual. The Con group was considered as reference 1, the other three groups were compared with the Con group; for MDA, the D-GH group was considered as reference 1, and the other three groups were compared with the D-GH group. Data are exhibited as mean ± standard error (SE) (n=15 mice). *P<0.05 versus the Con group; #P<0.05 versus the D-Gal group.
AD is a neurodegenerative disease with Aβ polymerization of melanin. Neuronal oxidative damage and loss of the forebrain cholinergic neurons have been observed in the disease [7]. The productions of reactive oxygen species (ROS), GSH-Px, SOD and MDA serve as biomarkers for lipid peroxidation. GSH-Px is one of the most widely distributed peroxide decomposition enzymes [8]. Polyunsaturated lipids are degraded to form MDA by ROS. MDA is a naturally occurring oxidative stress biomarker. SOD is a free radical scavenger and is widely distributed in biological tissue. It can remove O2 (superoxide anion free radicals), which is cytotoxic, can peroxidize lipids, damage the cell membrane, cause inflammation, tumors and autoimmune diseases, and may prompt bodily aging [9]. SOD can improve human immunity and anti-aging, reduces fatigue, adjusts female physiological cycles and delays menopause
As shown in Figure 3, for both the hippocampus and cortex, Aβ expressions of the D-Gal, D-GL and D-GH groups were significantly higher than that of the Con group; the D-GH group was significantly higher than the D-Gal group in the hippocampus; and both the D-GL and D-GH groups were significantly higher than the D-Gal group in the cortex.
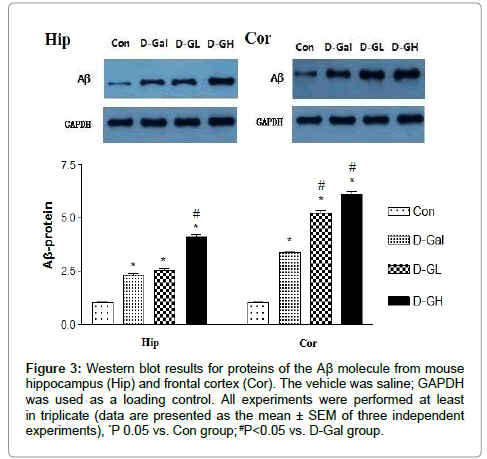
Figure 3: Western blot results for proteins of the Aβ molecule from mouse hippocampus (Hip) and frontal cortex (Cor). The vehicle was saline; GAPDH was used as a loading control. All experiments were performed at least in triplicate (data are presented as the mean ± SEM of three independent experiments), *P 0.05 vs. Con group; #P<0.05 vs. D-Gal group.
Aβ is one of the most important peptides produced from amyloid precursor protein (APP). Amyloid plaques are closely related to Alzheimer's disease. Evidence suggests that Aβ is a multifunctional and high level expressive peptide with significant non-pathological activity [10]. As the main constituent of brain parenchymal and vascular amyloid, it induces neurotoxicity and cerebrovascular lesions. Animal studies have shown that the absence of Aβ does not lead to any loss of physiological function, but the general function of Aβ is still not well known [11]. Potential actions of Aβ include activation of kinase enzymes, protection against oxidative stress, and regulation of cholesterol transport. Recent research suggests that soluble oligomeric forms of the peptide may be causative agents in the development of Alzheimer's disease [12]. A number of genetic, cell biology, biochemical and animal studies have supported the concept that Aβ plays a central role in the development of AD [13].
As shown in Figure 4, for both the hippocampus and cortex, NEP expression in the D-Gal, D-GL and D-GH groups was significantly lower than that in the control group; and expression in the both D-GL and D-GH groups was significantly lower than that in the D-Gal group.
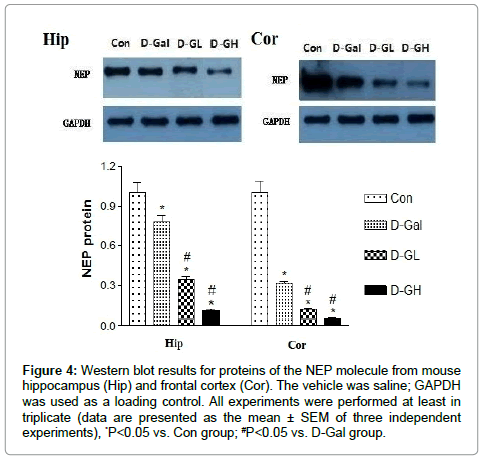
Figure 4: Western blot results for proteins of the NEP molecule from mouse hippocampus (Hip) and frontal cortex (Cor). The vehicle was saline; GAPDH was used as a loading control. All experiments were performed at least in triplicate (data are presented as the mean ± SEM of three independent experiments), *P<0.05 vs. Con group; #P<0.05 vs. D-Gal group.
NEP is a zinc-dependent metalloprotease enzyme that degrades a number of small secreted peptides, most notably the Aβ peptide of which abnormal folding and aggregation in neural tissue has been implicated as a cause of AD. Synthesized as a membrane-bound protein, the NEP ectodomain is released into the extracellular domain after it has been transported from the Golgi apparatus to the cell surface. NEP-deficient knockout mice show both Alzheimer's-like behavioral impairment and amyloid-beta deposition in the brain, providing strong evidence for the protein's association with the Alzheimer's disease process [14]. Because NEP is thought to be the rate-limiting step in beta amyloid degradation, it has been considered as a potential therapeutic target; compounds such as the peptide hormone somatostatin have been identified as agents that increase the enzyme's level of activity. One hypothesis for the strong relationship between the incidence of AD and age focuses on the declining production of somatostatin in the brains of elderly people; this depresses the activity of NEP and promotes aggregation of unprocessed beta amyloid [15]. Declining NEP activity with increasing age may also be explained by oxidative damage which is known to be a causative factor in AD; higher levels of inappropriately oxidized NEP have been found in Alzheimer's patients when compared to cognitively normal elderly people [16].
Figure 5 shows that, for both the hippocampus (Hip) and cortex (Cor), β secretase (BACE1) expression in the D-gal, D-GL and D-GH groups was significantly higher than that in the Con group; and expression in both the D-GL and D-GH groups was significantly higher than that in the D-gal group.
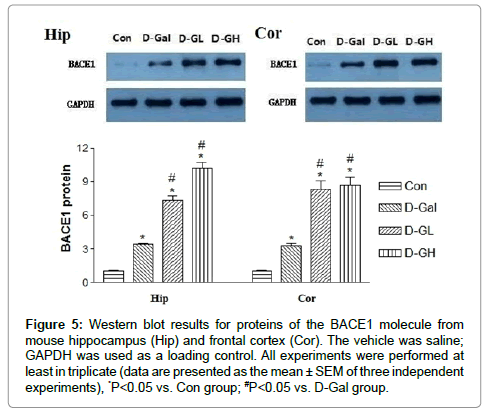
Figure 5:Western blot results for proteins of the BACE1 molecule from mouse hippocampus (Hip) and frontal cortex (Cor). The vehicle was saline; GAPDH was used as a loading control. All experiments were performed at least in triplicate (data are presented as the mean ± SEM of three independent experiments), *P<0.05 vs. Con group; #P<0.05 vs. D-Gal group.
Accumulation of disease-related proteins is characteristic of neurodegenerative diseases. BACE1, which initiates the generation of Aβ, is increased in the AD brain; however, the mechanism for BACE1 accumulation in Alzheimer's disease is largely unknown. In this study, we found that the intake of edible salt regulated the level of BACE1 protein. BACE1 levels were increased in response to Aβ or apoptosis. Furthermore, increased BACE1 expression facilitated APP being processed by the β-secretase processing pathway rather than the α-secretase pathway, thus leading to more Aβ production. Our study provided new insights into how alteration of BACE1 expression and β-secretase cleavage site selection could contribute to the pathogenesis of AD and the pharmaceutical potential of modulating BACE1 expression and its cleavage site selection.
The cellular levels of BACE1, the rate-limiting enzyme for the generation of the AD amyloid Aβ, are tightly regulated by two ERbased acetyl-CoA lysine acetyltransferases: ATase 1 and ATase 2 [17]. We found that both acetyltransferases were expressed in neurons and glial cells, and up-regulated in the brains of AD patients. We also identified the first and second generation compounds that inhibited ATase 1/ATase 2 and down-regulated the expression levels as well as the activity of BACE1. The mechanism of action involved competitive and non-competitive inhibition as well as the generation of unstable intermediates of the ATases that underwent degradation.
APP and presenilin mutations cause early-onset familial Alzheimer's disease (FAD). Some FAD-based mouse models have produced amyloid plaques, but others have not [18]. Aβ deposition can be manifest as compact and diffuse plaques, but it is unclear why the same Aβ molecules aggregate in different patterns. Is there a basic cellular process governing the pathogenesis of Aβ plaques? We showed that, in some FAD mouse models, compact plaque formation is associated with the progressive axonal pathology inherent in the increased expression of BACE1, the enzyme initiating the amyloidogenic processing of APP. Inhibition of BACE1 to prevent brain Aβ peptide formation is a potential disease-modifying approach to the treatment of Alzheimer's disease [19]. Despite over a decade of efforts, the identification of brain-penetrant BACE1 inhibitors that substantially lower CNS Aβ levels following systemic administration remains challenging. In this study we described the structure-based optimization of a series of brain-penetrant BACE1 inhibitors derived from an iminopyrimidinone scaffold
ES can enhance D-gal-induced dementia as demonstrated by significantly decreased memory and significantly prolonged escape time and distance to the platform by mice. ES also significantly increased the expression of Aβ and BACE1 and significantly decreased NEP.
In conclusion, excessive salt intake can regulate the protein expressions of Aβ, BACE1 and neprilysin to enhance D-gal-induced dementia.
Acknowledgment
This work was supported by the Natural Science Foundation of Inner Mongolia (No. 2011MS1167).
Conflict of interest
We have no conflict of interest in the manuscript files.
References
- Li C, Zhao R, Gao K, Wei Z, Yin MY, et al. (2011) Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer's disease.Curr Alzheimer Res 8: 67-80.
- Prakash A, Kumar A (2013) Pioglitazone alleviates the mitochondrial apoptotic pathway and mito-oxidative damage in the d-galactose-induced mouse model. Clin Exp Pharmacol Physiol 40: 644-651.
- Zhang Z, Wang X, Zhao M, Yu S, Qi H (2013) The immunological and antioxidant activities of polysaccharides extracted from Enteromorphalinza. Int J Biol Macromol 57: 45-49.
- Li M, Chapman S, Agho K, Eastman CJ (2008) Can even minimal news coverage influence consumer health-related behaviour? A case study of iodized salt sales, Australia.Health Educ Res 23: 543-548.
- Teas J, Pino S, Critchley A, Braverman LE (2004) Variability of iodine content in common commercially available edible seaweeds.Thyroid 14: 836-841.
- Minami A, Matsushita H, Horii Y, Ieno D, Matsuda Y, et al. (2013) Improvement of depression-like behavior and memory impairment with the ethanol extract of Pleurotuseryngii in ovariectomized rats. Biol Pharm Bull 36: 1990-1995.
- Row BW, Kheirandish L, Cheng Y, Rowell PP, Gozal D (2007) Impaired spatial working memory and altered choline acetyltransferase (CHAT) immunoreactivity and nicotinic receptor binding in rats exposed to intermittent hypoxia during sleep. Behav Brain Res 177: 308-314.
- Cavas L, Arpinar P, Yurdakoc K (2005) Possible interactions between antioxidant enzymes and free sialic acids in saliva: a preliminary study on elite judoists. Int J Sports Med 26: 832-835.
- Zou Y, Leu D, Chui J, Fike JR, Huang TT (2013) Effects of altered levels of extracellular superoxide dismutase and irradiation on hippocampal neurogenesis in female mice. Int J Radiat Oncol Biol Phys 87: 777-784.
- Li W, Yu J, Liu Y, Huang X, Abumaria N, et al. (2013) Elevation of brain magnesium prevents and reverses cognitive deficits and synaptic loss in Alzheimer's disease mouse model.J Neurosci 33: 8423-8441.
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, et al. (2011) Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl AcadSci USA 108: 5819-5824.
- Pomara N, Bruno D, Sarreal AS, Hernando RT, Nierenberg J, et al. (2012) Lower CSF amyloid beta peptides and higher F2-isoprostanes in cognitively intact elderly individuals with major depressive disorder. Am J Psychiatry 169: 523-530.
- Kaushik DK, Basu A (2013) A friend in need may not be a friend indeed: role of microglia in neurodegenerative diseases. CNS NeurolDisord Drug Targets 12: 726-740.
- Coppey L, Davidson E, Lu B, Gerard C, Yorek M (2011) Vasopeptidase inhibitor ilepatril (AVE7688) prevents obesity- and diabetes-induced neuropathy in C57Bl/6J mice. Neuropharmacology 60: 259-266
- Fischer HS, Zernig G, Hauser KF, Gerard C, Hersh LB, et al. (2002) Neutral endopeptidase knockout induces hyperalgesia in a model of visceral pain, an effect related to bradykinin and nitric oxide. J MolNeurosci 18: 129-134.
- Ni R, Gillberg PG, Bergfors A, Marutle A, Nordberg A (2013) Amyloid tracers detect multiple binding sites in Alzheimer's disease brain tissue. Brain 136: 2217-2227.
- Ding Y, Ko MH, Pehar M, Kotch F, Peters NR, et al. (2012) Biochemical inhibition of the acetyltransferases ATase1 and ATase2 reduces ß-secretase (BACE1) levels and Aß generation. J BiolChem 287: 8424-8433.
- Cai Y, Zhang XM, Macklin LN, Cai H, Luo XG, et al. (2012) BACE1 elevation is involved in amyloid plaque development in the triple transgenic model of Alzheimer's disease: differential Aß antibody labeling of early-onset axon terminal pathology. Neurotox Res 21: 160-174.
- Stamford AW, Scott JD, Li SW, Babu S, Tadesse D, et al. (2012) Discovery of an Orally Available, Brain Penetrant BACE1 Inhibitor that Affords Robust CNS Aß Reduction. ACS Med Chem Lett 3: 897-902.
Citation: Gong GH, Xi Wei C, Wang D, Bian M, Bao SY, et al. (2015) Excessive Intake Salt Contribution to Cognitive Impairment in Mice. J Mol Pharm Org Process Res 3: 124 DOI: 10.4172/2329-9053.1000124
Copyright: ©2015 Gong GH, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 15567
- [From(publication date): 8-2015 - Jul 12, 2025]
- Breakdown by view type
- HTML page views: 10949
- PDF downloads: 4618