Investigation of Invasive Species Corbicula Fluminea and Microbial Contents of the American River
Received: 03-Apr-2023 / Manuscript No. jee-23-93105 / Editor assigned: 05-Apr-2023 / PreQC No. jee-23-93105 (PQ) / Reviewed: 19-Apr-2023 / QC No. jee-23-93105 / Revised: 21-Apr-2023 / Manuscript No. jee-23-93105 (R) / Published Date: 28-Apr-2023 DOI: 10.4172/2157-7625.1000390
Abstract
In the past century, pollution and invasive species have plagued North American freshwater ecosystems. Recent reports state that fecal contamination has resulted in Escherichia coli outbreaks in Sacramento’s American River. The Asiatic clam (Corbicula fluminea) has devastated North American freshwater ecosystems for its great filtering abilities that outclass native species. This study was conducted to better understand the effects C. fluminea on freshwater ecosystems and the microbial contents of the American River. Among coliform bacteria, E. coli were estimated to comprise roughly 4.3% of detected coliforms in sample river water. Western blotting was conducted to successfully detect P-gp among gill tissue proteins in tested clams, which are associated with the tolerance of C. fluminea to polluted conditions due to their function in xenobiotic resistance. P-gp was detected at different relative abundancies in two Asiatic clams.
Introduction
Freshwater and marine ecosystems worldwide have been subject to higher levels of pollution and contamination since the beginning of the Industrial Revolution. Rivers and lakes across North America have faced such issues as the introduction of invasive species and effluent contamination due to sewage and agricultural runoff. Since its introduction in 1938 [1], the Asiatic clam (Corbicula fluminea) is among the most successful of invasive species to appear in freshwater rivers across the North American continent. Coupled with a fast reproductive rate and tolerance to different xenobiotics, C. fluminea has played a major role in transforming both biotic and abiotic factors of local aquatic environments due to its great biofiltration capacity. Effluent contamination has also contributed greatly to the transformation of the freshwater ecosystems like the American River in the greater Sacramento region. After reports of Escherichia coli outbreaks at the American River, E. coli and other coliforms have been detected in both river water samples and C. fluminea gill tissue extracts. Detection of E. coli in such river water is especially alarming due to the dependence of local communities on the American River for drinking water and agricultural purposes. Ingestion of E. coli by humans may induce such symptoms of varying severity as diarrhea, abdominal cramping, or nausea and vomiting that depend upon the strain [2].
C. fluminea and other bivalves are characterized by a general anatomical structure consisting of a foot used for transportation, two mantles that each generate a calcium carbonate-based shell [3,4], gills for filtration of contents dissolved in aqueous solvent, adductor muscles to close the two shells together, and other visceral mass that includes a heart and a digestive gland.2 Asiatic clams intake water from surrounding aqueous environment using their incurrent siphon so that the gills can filter oxygen and food particles. Unfiltered contents and waste products are excreted in water that is expelled out of the excurrent siphon [5].
While it shares many homologous features with other bivalves in the American River, the success of C. fluminea can be attributed to such features as a great biofiltration capacity, multixenobiotic resistance, and a rapid reproductive rate.
The ability of bivalves like C. fluminea to filter feed is attributed to the trapping of microbes and other food particles in their cirri, small fused cilia found in gill tissue capable of creating water vortexes or simply obstructing particles [6]. Despite its relatively small size when compared to native bivalve species [7], the broad and efficient filtration abilities of C. fluminea have allowed it to outcompete other bivalves and native species for nutrient-rich particles in surrounding water, like photosynthetic microbes with an important role as ecosystem engineers in the American River. Colonies of C. fluminea are capable of transforming surrounding aquatic environments by filtration. In a study where different bivalves were to filter salts from artificial pond water, C. fluminea were observed to a clearance rate more than triple that of zebra mussel (Dreissena polymorpha) specimens after controlling for gill surface area. It has also been reported that Asiatic clams can filter spherical algae particles as big as 50 μm in diameter and other particles as small as 1 μm. The high capacity of the Asiatic clam to bioaccumulate substances dissolved in surrounding aqueous solution make it a great candidate for investigating the health of local aquatic environment.
In addition to their ability to efficiently filter microbes and inorganic matter, some of the success of C. fluminea as an invasive species may be attributed to its great tolerance to environmental conditions. Bivalves like C. fluminea and the eastern oyster (Crassostrea virginica) are reported to exhibit multixenobiotic resistance (MXR) [8, 9]. P-glycoproteins (P-gps) are a part of a family of ATP-binding cassette (ABC) transporter proteins that have been directly associated with an MXR phenotype in bivalves and other aquatic orgnaisms. Cell membrane P-gp transporters in bivalves have a function of pumping out both organic and inorganic xenobiotics, requiring hydrolysis of 2 or 3 ATP molecules per molecule of substrate transferred. Resistance to such xenobiotics as cadmium and rhodamine B dye has been exhibited by bivalves with cell membrane P-gp expressed in gill tissue.
The purpose of this study was to investigate C. fluminea as a contributor to the changing environment of the American River. Asiatic clams, river water, and river bed soil samples were harvested from the American River in order to better understand their implication for the health of the American River. The river water sample was tested to estimate the concentrations of E. coli and other coliforms in the river. The aqueous contents of the soil sample and subsequent dilutions were plated to estimate the original cell density of agar-friendly microbes. A starved single Asiatic clam was placed in aqueous environment only containing a specific class of microbes; this was done to estimate the rate at which C. fluminea can filter E. coli and the rate at which it can filter algae. Spacing between rows of cirri was measured in different C. fluminea specimens in hopes of better understanding the influence of such spacing on filtration rates. The mitochondrial gene for cytochrome C oxidase subunit I in Asiatic clam specimens was amplified using PCR so it could be spread on agarose gel and sequenced; it was intended that the C. fluminea COI gene could be compared to genome segments in a database to verify whether the specimen was an Asiatic clam. By means of Western blotting, Asiatic clam gill tissues were inspected for traces of P-gp. It was hypothesized in this study that Asiatic clams subjected to higher concentrations of xenobiotics would have greater detectable traces of P-glycoprotein.
Materials and Methods
Field collection
All samples collected for this study were obtained from a small site of the shore of the American River in Folsom, CA off Blue Ravine Road. Waders were worn over normal clothing to protect wardrobe. Three 200-mL conical tubes and one plastic container containing nutrient pellets for microbes were placed into a carrying bag that was held by one researcher. The other researcher carried a shovel that would be used to dig up samples. After entering river water that was about three to four feet deep, three live Corbicula fluminea specimens were retrieved from the soil and stored in a single conical tube filled with local river water for later use in this study. All C. fluminea specimens used in this study would be obtained at this location. The nutrient containing container was just barely submerged in the river water so that a river water sample could be retrieved without loss of the nutrients in the container. These nutrients were placed inside to feed any live microbes in the water. A soil sample rich in soil water was shoveled from the surface of the river bed and placed into a second conical tube. These samples reached a research lab about forty minutes later. The clams were placed in a goldfish tank filled with tap water before being used at least one week later. The soil and water samples were incubated at 37°C and used two weeks later.
Experimental design
This study consisted of a series of experiments and other procedures conducted to understand Corbicula fluminea and its abilities in greater detail. First, three C. fluminea specimens were each extracted for samples of gill and mantle that were then stored in a freezer for at least four months before being retrieved a gain. The river water sample was inspected for its concentration of coliform bacteria using Coliplate™ trays and a most probable number (MPN) chart. The moist soil-water mixture was diluted three times and each dilution was cultured to measure for the concentration of microbes that could grow in nutrient agar. A single Asiatic clam specimen was then used in an experiment in which its rate of filter feeding of microbes in water only mixed with either Escherichia coli bacteria or algae broth culture. After the second filter feeding test was completed, the clam was dissected for its gill tissue. The gill tissue was observed under a microscope for observation of its cirri. This clam and five other clams would be compared for their spacing between adjacent lines of cirri. Two of the three stored C. fluminea mantle tissue specimens were used for extraction of Asiatic clam DNA where only a segment of mtDNA was amplified by PCR, purified, and observed using agarose gel electrophoresis.
Dissections
All clam tissue specimens used in this study were obtained using the same technique. A dissection kit containing forceps, scissors, and a scalpel was used to the dissect the clam of focus. Clams were dissected over a lab tray while a cut-resistant glove was worn over the non-dominant hand. Additional instruments present at the laboratory station included a calibrated balance, six 1.5-mL microcentrifuge tubes, and an ice bath.
All live clams dissected in the lab had their two shells closed tightly by their adductor muscles, making it difficult to peek at components of the clam deep to the mantle. Using the dominant hand, a scalpel was inserted into siphons just barely visible on the dorsal side of the clam. The scalpel and scissors were used to slowly cut around the perimeter of the tissue just deep to the most lateral sections of the clam shells. This was done to cut through the adductor muscles in hopes of exposing the internal anatomy of the clam. After refracting the superior shell of the clam back, it became easier to observe the asymmetrical internal anatomy of the clam. The foot of the clam generally occupied most of the ventral internal mass. Gill tissue was extracted from a lateral site of internal mass proximal to the site of the initial incision into the siphons of the clam. Mantle tissue was also extracted. Collected gill tissue specimens were between 20 milligrams and 90 milligrams. Collected mantle tissue specimens were less than 50 milligrams.
To properly measure the mass of each sample, 1.5-mL microcentrifuge tubes were first placed on a calibrated balance. The balance was then tared to only account for the mass of any substances placed inside of the tubes. The tissue specimen of interest was extracted using scissors to cut away and forceps to transfer the specimen inside of the microcentrifuge tube. The tube was then placed back on the balance so that the tissue specimen mass could be recorded. These tubes were immediately placed onto the ice bath afterwards to better preserve the specimens. If tissue specimens were not to immediately be used, they were stored in a freezer at 4°C.
Soil microbial detection sample collection
As described in the “Field collection” section, river water and soils samples were obtained from the American River at a site in Folsom, CA. River water was obtained by just barely submerging a nutrientcontaining vessel in the river so that no nutrient was lost. Soil specimens moistened by river water were collected by shoveling the surface of the bed of the river and placing it into a conical tube. These samples were then incubated at 37°C in a lab for about 334 hours (about fourteen days) before being retrieved for analysis.
Soil sample procedure
The soil sample previously described was retrieved from the incubator and taken to the laboratory for analysis of its concentration of microbes that can grow in standard nutrient agar. In preparation for this analysis, the following materials were obtained: three petri dishes containing nutrient agar, P1000 and P200 micropipettes with appropriate sterilized tips, 1.5-mL microcentrifuge tubes, a supply of sterilized deionized water, and sterilized cotton swabs. Samples were labeled using a grease pencil.
Three microcentrifuge tubes were labeled as either “100,” “10- 1” and “10-3” for identification of their future contents, dilutions of the soil water in the previously incubated conical tube. An original soil water solution sample and two dilutions would be placed in the microcentrifuge tubes. First, 1000 μL of soil water solution was extracted from near the top of the contents of the conical tube and deposited into the 100 tube using the P1000 micropipette. The tube was immediately shaken to mix its contents. A dilution of the soil sample was prepared in the 10-1 tube using 100 μL of the soil water solution in the 100 tube and 900 μL of deionized sterile water. This tube was also shaken to homogenize its contents. 100 μL of the dilution was extracted using the P200 micropipette and placed into the 10-2 tube. 900 μL was also placed in the 10-2 tube before the tube was mixed to homogenize the mixture to prepare a dilution of the original prepared soil solution in the 100 tube.
To prepare for the culturing of the prepared soil solutions and dilutions, the bottoms of three nutrient agar-containing petri dishes were labeled to describe their future contents. The labels specifically included the same names as the tubes containing the different soil mixtures. A sterile swab was carefully submerged in the 100 tube containing the original soil solution and immediately spread across the surface of the agar of the 100 plate. This process was repeated using the 10-2 and 10-3 tubes and the prepared and dilutions respectively. The plates were then sealed using electrical tape and incubated at 37°C for about 166 hours before being retrieved again.
The plates were retrieved again for inspection of colonies. Each plate was observed for the number of colony forming units (CFUs) visible at the surface of the agar. A picture of each plate was taken and used to count for CFUs. The microbe concentration of each soil solution was calculated using original cell density (OCD) and its respective equation:
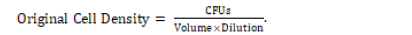
It should be noted that the OCD was calculated specifically for the original prepared soil solution in the 100 tube by considering how diluted a specific cultured dilution was.
Water sample procedure
During the same period in which the soil analysis was performed, an analysis of the river water was conducted to estimate the concentration of detectable coliform bacteria and the concentration of Escherichia coli in the American River. The river water sample in its nutrient-containing vessel was retrieved from the incubator. An unused Coliplate™ microplate, Kimwipe, and sterile disposable 1-mL pipettes were obtained in preparation for this segment of the study. A single pipette was used to transfer river water to each of the first twelve of sixty-four wells. It proved to be more effective and efficient when river water was poured directly over the microplate to fill all wells. Excess water was wiped off using Kimwipes. The microplate was then placed into an incubator at 37°C for about 24 hours. Although the microplates were designed for inspection after twenty-four hours of incubation, the plates could not be immediately observed and were thus placed in a freezer at 4°C for about 124 more hours in hopes of inhibiting further growth by microbes. Unfortunately, microbes later appeared a greater frequency than intended.
The microplates were retrieved from the freezer for inspection of well color. If a well exhibited a blue color under standard lighting standard for a college undergraduate biology laboratory, that would imply that coliform bacteria have grown in the well. If a such a blue well shines a bright fluorescent blue under ultraviolet light, that would suggest the growth of E. coli in the well. The microplate was first inspected under previously described standard lighting conditions against a white background to count for blue wells. The microplate was transferred to a dark room and set upon a black table. An ultraviolet light was shined upon the microplate, and the quantity of fluorescent blue wells was counted. Estimates of the coliform concentration and E. coli concentration in the prepared river water solution were obtained by matching the respective number of wells counted to a value for the most probable number (MPN), an estimated number of CFUs per 1 milliliter of solution, on a chart provided with the Coliplate™ microplate.
Water sample data analysis
As described in the two previous subsections, estimates were gathered for different concentrations of microbes in the river water and the prepared soil solution. The original cell density (OCD) of the water in the American River was calculated as an estimate for the concentration of microbes that can grow in nutrient agar. The three estimates for OCD were then compared to make inferences about the contents of the American River. A most probable number (MPN) chart provided by Bluewater Biosciences Inc. [5] was used in estimating the number colony forming units (CFUs) in the river water sample collected from the American River. Different MPN values were obtained for the concentration of coliform bacteria and the concentration of E. coli, a single species of coliform bacteria, in the water in CFUs per milliliter of solution.
Invertebrate feeding subjects and controls
A continuous series of experiments were conducted in which a single Asiatic clam was monitored for its rate for filter feeding of E. coli and green algae respectively. In preparation of this experiment, the clam was starved to a point of hunger by being placed in 200 milliliters of distilled deionized water without being fed. The intent of this decision was to minimize the risk of confounding due to the presence of other microbes and to encourage the clam to use its siphons to filter as much as possible to satisfy dietary needs. Additional materials required for this experiment included prepared E. coli broth culture, green algal broth culture, disposable 1-mL pipettes, a supply of distilled water, a microplate, and a spectrophotometer.
Invertebrate feeding procedure
The first experiment to be conducted was that in which algal filter feeding rate of the Corbicula fluminea specimen was measured. All solution samples would be deposited into a single well of a microplate containing at least 32 wells. Two 2.0-mL samples of distilled water were first deposited into the first three wells of the microplate, “A1,” “B1,” and “C1,” to later function as blanks for the spectrophotometer. Only three droplets of the algal broth were dropped into the water the clam was submerged in. Another pair of 2.0-mL sample of the solution was carefully extracted and placed into wells “D1” and “E1.” After fifteen minutes, two additional 2.0-mL extractions were placed into wells “F1” and “G1.” With each successive time interval of fifteen minutes, pairs of 2.0-mL samples of the aqueous mixture surrounding the clam were extracted and transferred into the next two wells until there were two samples for every fifteen-minute time interval up about 120 minutes after the addition of algal broth. After the microplate was filled with each of the collected solution samples, the well was placed into a spectrophotometer set to a frequency of 550 nm of electromagnetic radiation. The first three wells containing distilled water functioned as a blank. The absorbance of all the water samples from the brothcontaining aqueous mixture surrounding the clam was observed. The pairs of absorbance values for a given point in time after addition of algal broth were used in preparing a scatterplot that depicted the relationship between absorbance versus minutes of time. The negative slope of the least squares regression line was used as an estimate of the algal feeding rate of the C. fluminea specimen.
This procedure was nearly replicated in a second experiment with intentions of estimating the filter feeding rate of E. coli by the same Asiatic clam used in the previous filter feeding experiment. The clam same clam was starved again, and E. coli broth culture was prepared for the experiment. An unspecified amount of sterile E. coli broth culture was first centrifuged at 3000 rpm for five minutes. All available brown liquid was used while pellets formed at the bottom of the centrifuge tube were ignored. 1 mL of sterile dechlorinated water was added to the brown liquid for every 50 mL of mixture and mixed to resuspend the bacteria. The procedure described for the algal feeding rate experiment was then exactly replicated using the new E. coli broth culture instead of the algal broth culture. Using the same spectrophotometer set to a frequency of 550 nm, absorbance values were obtained for each pair of 2.0-mL solution samples. These absorbance values were used to make another scatterplot depicting the relationship between absorbance of the solution versus the number of minutes after the prepared E. coli broth culture was added to the water containing the clam. The negative slope of the least squares regression line was used to estimate the filter feeding rate of E. coli by the same Asiatic clam.
Invertebrate feeding data analysis
The main objective of both filter feeding experiments was to obtain an estimate for the filter feeding rate of either green algae or Escherichia coli by an Asiatic clam using absorbance values of the clam’s surrounding solution after specific points in time after addition of green algal broth. To the green algal feeding rate by the clam, a series of twenty-seven ordered pairs representing the twenty-seven collected solution samples were plotted to generate a scatterplot that depicts the relationship between the number of minutes after addition of the appropriate broth solution and absorbance of 550 nm electromagnetic radiation by the extracted sample of solution surrounding the clam at the respective point in time. Extreme outliers that deviated by at least 1.5 standard deviations from provided estimate by the original least squares regression line were omitted. The least squares regression line of all points excluding previously described extreme outliers was generated on Microsoft Excel for the values of its slope. The negative slope (the opposite of the slope) was used as an estimate or the feeding rate. The estimated feeding rate of E. coli and the estimated feeding rate of green algae by the C. fluminea specimen were then inspected for validity and compared to each other.
Cirri spacing sample preparation
The spacing between cirri of C. fluminea was analyzed in attempts to generate a reasonable estimate of the average distance between adjacent rows of cirri in C. fluminea. Materials used in this section included a dissection kit, a dissection tray, a plastic slide, and a complimentary plastic square for the slide.
Six live Corbicula fluminea specimens were subject to the conditions of both the algal filter feeding experiment and the E. coli filer feeding experiment, including the one used in the two experiments in the previous section “Invertebrate Feeding.” These six clams were each dissected for a sample of gill tissue to be observed under a microscope. Each clam was dissected using the procedure described in the “Dissections” subsection. Each retrieved tissue specimen was placed onto a plastic side and flattened using a thin plastic square with a width only a bit smaller than that of the slide.
Gill tissue imaging
A teaching microscope capable of digital photography was used to inspect the gill tissue slides for cirri spacing. A hemocytometer was placed under the microscopic at an unspecified magnification for the duration of the entire procedure. A picture was taken in which an entire patch of 0.05 mm squares were visible. Pictures were taken of each gill tissue slide at the same magnification for cirri to be observed. A computer program called “ImageJ” to calibrate a digital ruler to the pixel count of image depicting the hemocytometer’s patch of 0.05 mm squares. This digital ruler was then used to measure the length of cirri spacing in the images of the six slides. Cirri spacing was compared within and between samples to yield a reasonable estimate of the average cirri spacing among the six images C. fluminea tissue samples.
Cirri spacing data analysis
Each image of C. fluminea gill tissue was inspected for three different measurements of spacing between the same rows of cirri and determine the average of these measurements. For an image of the same clam specimen, this was done with three different pair of adjacent rows. A grand mean value for the series of measurements for the three inspected paired rows was determined for each clam tissue specimen. A box plot was generated depicting the spread of cirri spacing values for the data set of grand mean values. A mean value for the grand mean of cirri spacing for each clam was calculated and used as an estimate for the average cirri spacing in tested C. fluminea specimens.
DNA Barcoding sample preparation
DNA barcoding of C. fluminea was conducted using DNA extracted from two of the frozen mantle tissue specimens prepared according to the “Dissection” section about four months prior to enactment of the following described procedure. For the process of extracting DNA from the mantle tissue, the following materials were used: a 1 M of Tris solution (with a pH of about 7.4), a 5 M solution of sodium chloride, a 0.5 M solution of EDTA, 100 percent liquid ethanol, double distilled water (ddH2O), micropipettes with appropriate sterilized tips, disposable 10-mL graduated pipettes, an ice bath, silica resin, wash buffer, a microcentrifuge, 1.5-mL microcentrifuge tubes, plastic pestles, a vortex machine, and sterile deionized water.
A lysis buffer needed for DNA extraction from the mantle tissue was first prepared. In a 20-L beaker, 2340 μL of double distilled water was mixed with 2500 μL of 100% ethanol. 100 μL of 1 M Tris solution, 50 μL of 5 M sodium chloride solution, and 10 μL of 1.0 M EDTA solution were each added to ted water-ethanol liquid mixture. The contents were thoroughly mixed to yield a lysis buffer solution containing the following reagent concentrations: 20 mM Tris, 50 mM NaCl, 1 mM EDTA, and 50% ethanol (by volume).
The previously described frozen C. fluminea mantle samples were first identified and then transferred to an ice bath to decrease risk of denaturing DNA. Although three mantle samples were prepared, only two were used for the rest of the procedure. 100 μL of the prepared lysis buffer was added to the microcentrifuge tubes of each of the two mantle samples. The lysis buffer solution is used to dissolve membranebound organelles like the nucleus or the mitochondrion. A different clean plastic pestle was used to homogenize the frozen mantle tissue while it was submerged in the lysis buffer. Grinding of the tissue with the pestle helps to break up cell membranes and other tough materials. The homogenized tissue samples in lysis buffer were then placed in a 55°C heat block for 30 minutes. Immediately afterwards, the samples were transferred a second heat block at 100°C for eight minutes. The heated samples were then placed into a microcentrifuge set at 14,000 rpm for 5 minutes. This first centrifuge should pellet out cellular debris at the bottom of the 1.5-mL microcentrifuge tube housing the samples while the genomic DNA, proteins, and other water soluble materials remained in the supernatant. Two new microcentrifuge tubes were obtained and labeled with sample IDs of each gill tissue sample with an additional “S1” for “supernatant 1.” For each sample, a P200 micropipette was then used to carefully transfer 100 μL of supernatant from the original tube now containing a pellet to the newly labeled “S1” tubes. The two original tubes were then disposed of.
50 μL of silica resin was added to each newly prepared “S1” tube. The contents of these tubes were homogenized by use of a vortex machine. Silica resin is a substance that binds to DNA, making a DNA-silica resin complex that is insoluble in the prepared lysis buffer. The “S1” tubes were centrifuged at 14,000 rpm for two minutes for formation of a new pellet consisting of silica resin and the genomic DNA. After extraction from the microcentrifuge, the supernatant was carefully removed from both “S1” tubes both by use of a micropipette and turning the tube 180° so that it could be upside down. Now, 500 μL of ice-cold (0°C) wash buffer was added to the “S1” tube pellet, and the tubes were soon vortexed to resuspend the silica resin. The “S1” tubes were centrifuged at 14,000 rpm for one minute to re-pellet the resin and DNA. The supernatant was carefully removed using a micropipette. This series of actions of adding wash buffer, mixing, centrifuging, and removing the supernatant was done to clean the tubes of any unwanted biomolecules while the DNA is still bound to the silica resin, which is insoluble to the wash buffer. The series of actions to remove unwanted biomolecules was promptly repeated a second time with both tubes. The tubes were then left with the lids opened for over one minute to let the pellets dry, and a hair dryer was used to speed up the process. 100 μL of deionized sterile water was added to each—now completely dried—pellet to resuspend the DNA in aqueous solution, because DNA is soluble to water while silica resin is insoluble to water. Next, the “S1” microcentrifuge tubes were placed into a 65°C for ten minutes. These tubes were then centrifuged one more time at 14,000 rpm for one minute to pellet the resin and elute any missing DNA from the resin. Two new 1.5-mL microcentrifuge tubes were obtained and labeled exactly as the “S1” microcentrifuge tubes were. 90 μL of each supernatant solution was transferred to their respective new, empty labeled tubes before being stored in a freezer at 4°C for about 166 hours.
Polymerase chain reaction
Polymerase chain reaction (PCR) was conducted to amplify the concentration of a specific sequence of C. fluminea mtDNA as specified in the “Introduction.” Specific DNA primers were ordered to promote replication of a 700 base pair section of the 1200 base pair gene for cytochrome C oxidase subunit I (COI), an important component of the electron transport chain. Solutions obtained for preparation of a “PCR master mixture” included 10 X deoxynucleotide triphosphates (dNTPs), 2 nM of primer mix, 10 X of PCR buffer, sterile double distilled water (ddH2O), and Taq DNA polymerase. All these substances were kept in an ice bucket while not being used. Samples amplified according to the following protocol included the two prepared C. fluminea DNA samples previously extracted from mantle tissue, a control DNA sample from C. elegans (a species of nematode), and a sample of pure sterilized water.
To begin preparation of the PCR master mixture, a 1.5-mL microcentrifuge tube was obtained and labeled “MM” as short for “Master Mix.” 25 μL of 10 X PCR buffer and 180 μL of ddH2O were first added to the “MM” tube. Then, 5 μL of dNTP solution and 25 μL of primer mix solution were added. 5 μL of Taq polymerase solution was added last. Four PCR-specific tubes were obtained and carefully labeled for their future contents, because the labels could have easily been removed while later subjected to intense heat. 24 μL of prepared PCR master mixture was added to each of the four PCR tubes. Afterwards, 6 μL of each DNA sample or control sample was added to its respective label tube.
All four tubes were transferred to a thermocycler, and the respective locations of the four tubes were recorded. The thermocycler was set to initially stay at a temperature of 94°C for one minute. After that, the thermocycler would go through 35 cycles of the three following steps: First, it would go to 95°C for 30 seconds. In this step, sample dsDNA molecules are being denatured by the heat. Second, the thermocycler would go to 55°C for 30 seconds. At this temperature, the DNA primers would just barely anneal to their intended complimentary sites. The third step would be the thermocycler staying at 65°C for 30 seconds, an optimal temperature for reasonable time to allow Taq polymerase to elongate the DNA primers before dsDNA is denatured again. After the 35 series of cycles were complete, the amplified DNA samples and control samples were transferred to a freezer at 4°C for about 166 hours.
Agarose gel electrophoresis
Gel electrophoresis was conducted to evaluate whether the number of base pairs in the DNA extracted from the C. fluminea mantle tissue has the number of base pairs that is expected, about 700 base pairs. 1.0% agarose was used in this gel electrophoresis procedure. Five wells were occupied with either NEB 100 bp Ladder, the amplified first “S1” C. fluminea DNA sample, the amplified second “S1” C. fluminea DNA sample, the amplified C. elegans DNA sample, and the PCR-processed negative control that originally just contained pure water.
To prepare the agarose gel, 25 mL of 1.0% agarose was poured into a casting tray whose ends were covered with white duct tape. A gel comb was place at one end of the gel, and air bubbles were popped using a micropipette. The agarose was allowed to set in a 20 minute period. Meanwhile, the PCR products were prepared for the gel electrophoresis. New 1.5-mL microcentrifuge tubes were labeled with those on the PCR tubes containing the four samples to be run on the agarose gel. Within each new microcentrifuge tube, 15 μL of PCT product was mixed with 3 μL of loading dye. When the agarose gel was done, it was placed into the center of a gel rig. The gel rig was filled with 1 X TBE buffer to the maximum fill line. The wells were then filled from left to right as follows: NEB 100 bp Ladder, the amplified first “S1” C. fluminea DNA sample, the amplified second “S1” C. fluminea DNA sample, the amplified C. elegans DNA sample, and the PCR-processed negative control. The gel rig was then plugged into a 100 V battery and allowed to run for 30 minutes. After the 30 minutes were over, the gel was placed into a UV transilluminator so that it could be imaged. After it was imaged, the gel was disposed of in a normal waste bin.
Gel electrophoresis data analysis
The intent of the gel electrophoresis of DNA samples was to verify that the amplified DNA sequence was the 700-base pair segment of the C. fluminea COI gene of interest for DNA barcoding. Using imaging software “Paint.net,” the previously obtained agarose gel image was analyzed for the bands in each lane and their relative distance from their well. Each band in the NEB 100-bp ladder was identified before proceeding further. Distance was measured using pixels of the gel image, because the imaged agarose gel was not measured for length before it was disposed of. Using ladder band size data, a semi log plot was generated where band length was on the distorted vertical axis and distance from the well in pixels was on the horizontal axis. Estimates of DNA segment length for bands in other lanes were derived using the regression line generated from the semi log plot.
DNA sequencing
One of the PCR-amplified DNA samples was used in DNA sequencing using both a “forward” primer and a “reverse” primer. Because our own laboratory did not have the resources available to sequence the cytochrome oxidase subunit I (COI) gene, our amplified DNA samples were sent to another laboratory with the resources needed for this process. In place of a protocol describing what done in our laboratory, a general overview of the DNA sequencing technique used will be described. Materials required for this technique include dideoxynucleotides (didNTPS), acrylamide gel, a heat resistant DNA polymerase, dNTPS, and additional materials like those previously listed in the “Polymerase chain reaction (PCR)” and “Agarose gel electrophoresis” procedures [10,11].
A microcentrifuge tube is first prepared to hold mixture containing the DNA sample of interest, a heat resistant DNA polymerase like Taq polymerase, a short primer complimentary to the DNA sequence of interest, standard dNTPs, and didNTPS. The mixture is heated to about 96°C to denature the hydrogen bonds between the DNA strands. The temperature is then lowered to 60°C to just allow the primer to bind specifically to its complimentary sequence embedded within a segment of sample DNA extract. After enough time has passed for primer annealing, the temperature of the mixture is raised to optimal conditions for the heat-tolerant DNA polymerase so it can add nucleotides to the 3’ end of the annealed primer. Because DNA polymerases cannot tell the different between dNTPs and didNTPs, complimentary sequences of differing lengths are created. Primer lengthening stops when a didNTPS is added, because dideoxynucleotides lack a hydroxyl group on carbon 3’, a site needed for formation of phosphodiester bond with another nucleotide’s 5’ end. The sample is then prepared for gel electrophoresis.
The contents of the microcentrifuge tube are placed into a well in polyacrylamide gel for the DNA strands of differing lengths to separated by gel electrophoresis. Next, the loaded gel is placed into a Sanger sequencing (“DNA sequencer”) machine that runs the gel. The machine then detects for emitted light of a characteristic visible spectrum wavelength for each dideoxynucleotide: Red for thymine, green for adenine, blue for cytosine, and yellow for guanine. Four graphs of representing each color wavelength are overlapped to yield an electropherogram, and each peak in a wavelength graph should indicate the detection of a didNTP. Good DNA sequencing results should yield non-overlapping peaks of different colors that can be used to determine the original DNA sequence. A computer program can then be used to determine the most likely dideoxynucleotide at a specific position in the gel lane occupied by the sequenced DNA. A series of the nitrogenous bases from these didNTPS can thus be interpreted as the original sequence of the DNA sequence, where the nucleotide on the didNTP of the shortest DNA strand is the 5’ end of the sequence. It should be noted that electropherograms generally represent the yellow wavelength detection for guanine as black to make it easier to see. In the IUAC system for nucleic acid codes, an entirely unidentifiable nucleotide is designated as “N” and a series of other codes are used to designate such other uncertainties as an unidentifiable pyrimidine being marked as “Y.”
DNA sequencing data analysis
The purpose of the DNA sequencing procedure was to yield electropherograms from the sequenced DNA from which a DNA sequence could be obtained. Two “.ab” files containing electropherograms were obtained from the other laboratory that sequenced the single PCR-amplified DNA sample using a forward primer and a reverse primer respectively. This section intends to describe how these two electropherograms were used to try verifying that the sequenced gene was the same as the gene expected, a 700- base pair segment of the C. fluminea cytochrome C oxidase subunit I (COI) mitochondrial gene. To assure that our primers were appropriate for our intended gene, we conducted a “Nucleotide BLAST” search using the Basic Local Alignment Search Tool (BLAST) to individual compare each of the primers (written in FASTA format) to sequenced DNA segments in the NCBI database. After receiving matches with sequences of the COI gene in different species, the forward primer and reverse primer “.ab” files were inspected in Finch TV, free software for electropherogram inspection, for their respective electropherograms and computer-assigned nucleotide sequences. Each sequence was trimmed modified to yield the best sequence data possible before being converted to “.seq” file format. Two more “Nucleotide BLAST” searches were conducted for each trimmed and modified DNA sequence. The forward primer-based sequence was first searched for in the NCBI database and no genome segments were matched to the sequence. This result was also true of the reverse primer. While there was intent to compare the obtained sequences to database sequences to yield an E-value statistic, no matches were found, and the rest of the intended procedure was aborted.
Protein expression sample preparation
Western blotting of Asiatic clam gill tissue was conducted in search of P-glycoprotein (P-gp), a membrane protein associated with multixenobiotic resistance. Gill tissue samples previously extracted from two C. fluminea specimens were tested for detection of P-gp. To prepare samples for SDS-PAGE and electroblot transfer procedures, membrane and intracellular proteins needed to be separated from other cellular contents. Laboratory instruments and other materials used in this sample preparation included gill tissue samples in microcentrifuge tubes, buffer K (a homogenizer buffer), a plastic pestle for 1.5-mL microcentrifuge tubes, sample bugger, 1.5-mL microcentrifuge tubes, micropipettes with sterile tips, and a grease pencil for labeling. Samples were kept in an ice bath for as much time as possible to minimize denaturation. Buffer K was prepared prior to sample preparation and included the following concentrations of reagents: 5 nM MgCl2, 5 nM NaH2PO4, 40 nM HEPES, 70 nM potassium glutamate, and 150 nM Sorbitol. Sample buffer used in this procedure contained glycerol, bromophenol blue, β-mercaptoethanol (BME), sodium dodecyl sulfate (SDS), and Tris-HCl. BME was important to break up disulfide bridges between proteins while SDS can apply negative charges to amino acid residue side chains. The application of these reagents in sample buffer to extracted proteins is important for controlling for protein folding and charge during SDS-PAGE, a variant of gel electrophoresis used for separating proteins solely based on charge.
Three gill tissue samples weighing no more about 80 milligrams were prepared for extraction of cellular proteins. First, buffer K was added to each sample at a volume of 50.0 μL per 10.0 grams of gill tissue sample. After buffer K was added to a sample, a new sterile plastic pestle was used to grind up the tissue to homogenize its contents. Each sample was left on the ice bath for about 1 minute after its tissue was grinded. Using a new sterile tip each time, each homogenized sample was transferred to a new microcentrifuge tube appropriately labeled for its respective source C. fluminea specimen. Under a fume hood, sample buffer was added to each sample at the same volume as the volume of buffer K initially added prior to homogenization. The samples were then vortexed to mix their contents. After their respective contents were thoroughly mixed, the three samples were placed into a heat block at 100°C for five minutes. This was done to disrupt the tertiary and quaternary structure of extracted proteins while sample buffer contents also acted to change the protein structure and apply negative charge. After the samples were heated, they were placed onto an ice block before their use in SDS-PAGE one week later.
Western blot procedure
The Western Blotting technique used in this study consisted of sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDSPAGE), electroblot transfer to PVDF membrane, and application of antibodies and chemiluminescent substrate. Materials gathered for the SDS-PAGE procedure included pre-cast polyacrylamide gel (~5% stacking gel, ~10% separating gel), a tetra cell, 1x running buffer, casting gaskets, loading tips for micropipettes, a power supply, a Western gel rig with inserts, filter paper, filter pad, a plastic scraper, 1x transfer buffer (in ice bath), a stir bar, a stir plate, an ice pack, a plastic tray, and a precast-gel cracker. Other materials for the electroblot transfer and chemiluminescent imaging included a PVDF membrane, Ponceau S solution, TBST solution (0.1% Tween), P-gp-specific primary antibody, secondary antibody, ECL substrate, a small plastic container, a graduated cylinder, sterile 15-mL conical tubes, forceps, a Bio-Rad transilluminator, a Bio-rocker, sealable plastic (bags), and a chemiluminescent pen.
SDS-PAGE of gill tissue protein extracts was conducted to separate proteins by size before the transfer of such proteins to PVDF membrane. To begin preparations for SDS-PAGE, the three gill tissue protein samples were heated by heat block at 95°C for about 5 minutes. Meanwhile the tetra cell was prepared so the pre-cast polyacrylamide gel was already sitting in the cell with its gel comb removed. SDSRunning Buffer was added to fill the clamping frame about halfway. The frame clamping frame was placed into the tank and left untouched for five minutes. Afterwards, additional running buffer was added to the inner chamber to a height where the wells were completely submerged in buffer. The outer chamber was filled to the “2 Gels” mark; a gel for another experiment was placed into the tetra cell alongside the gel for this experiment. 15μL of NEB 100 kDa ladder was added to the first well. The other wells had 30 μL of their respective contents (from wells 2 to 8 respectively): positive control sample (Caenorhabditis elegans DNA segment), negative control sample, gill tissue protein extract from clam F180305A1, gill tissue protein extract from clam F180305A2, 200 ppm CaCl2, and 100 ppm CaCl2. The tetra cell was attached to its power supply, which was soon set to 120 V for about 60 minutes. After those 60 minutes, the power supply was turned off so that the negatively charged bands would stop migrating to the cathode. The gel was later extracted from its plastic casing by use of the precast-gel cracker.
Electroblot transfer from polyacrylamide gel to PVDF membrane required the formation of a gel-membrane cassette “sandwich.” The sandwich was made in a plastic tray container with the black anode half of the gel container was entirely submerged into 1x transfer buffer. In this order from bottom to top, the sandwich was made: Black anode side of gel holder, pad, and filter paper, polyacrylamide gel with samples, PVDF membrane, pad, and clear cathode side of gel holder. After checking for air bubbles between the filter paper and membrane, the sandwich was closed. The sandwich was inserted into a cassette that could be placed into a tetra cell. In addition to the cassette, an ice pack and stir bar were placed into the tetra cell. While the connected to a power supply, the tetra cell received a 100-mA current for about 2 hours to allow for effective transfer of extract proteins. When the electroblot transfer was finished, the membrane was placed into a small plastic bag containing TBST that was soon sealed. The bag containing the membrane was set upon a moving Bio-Rocker for about 144 hours (6 days). The membrane was then extracted from its bag and set into a plastic tray containing enough TBST to submerge the membrane. The TBST in the original bag was poured out before Ponceau S was immediately poured into an unused plastic bag. Soon the membrane was transferred into the unused bag containing Ponceau S, and the new bag with the membrane was set upon the oscillating Bio-Rocker for an additional 22 hours.
Primary antibody incubation began with use of the chemiluminescent pen to label the protein ladder on the membrane. The membrane was blocked by placing the membrane into a small container with 10 mL of 5%BSA/TBST solution and letting it rock for about 1 hour. Forceps were used to remove the membrane from its original bag and then placing it into a new, unused plastic bag. 1 g/ μL solution of P-gp specific antibody was added to the bag containing the PVDF membrane. Any excess primary antibody was recycled. The bag containing primary antibody was first sealed and then incubated overnight (21 hours) at 4°C. The blot was later retrieved along with 15 mL of TBST for the first membrane wash. The membrane was carefully removed from its vessel before it was placed into an empty plastic container. The first TBST wash was performed by adding 15 mL of TBST solution to the membrane and placing it on the Bio-Rocker for 5 minutes. The TBST solution was disposed of after five minutes. After two additional TBST washes were performed, secondary antibody solution was added to the membrane and its container. The membrane was placed on the oscillating Bio-Rocker at room temperature for 60 minutes. After the secondary antibody was disposed of, three more TBST washes were performed. Chemiluminescent substrates were mixed separate from the membrane container, but the membrane had switched locations. In its new container, chemiluminescent substrate was only added after the membrane was moved. The chemiluminescent enzyme is covalently bound to secondary antibody, and this enzyme catalyzes a reaction that causes the fluorescence that can be detected by the membrane imaging device. To minimize risk of confounding, the substrates were mixed not too long before they were added to the membrane. Only in locations where enzyme is present, on the secondary antibodies located at the protein bands, would the substrates react to be fluorescent.
Western blotting data analysis
ImageJ was used to determine the fluorescence of each band to quantify their relative protein content. Each well was selected in the program at the sites of 150-kDa bands, where P-gp should have been detectable. Using available command prompts, a graph depicting fluorescence of the band at series of positions. To account for the fluorescence of the entire band, area under the curve of significant troughs was taken for each band and reported as “Chemiluminescence.” More fluorescent wells generally had greater P-gp content.
Results
A ColiplateTM microplate used for testing total coliform and E. coli concentrations in the American River (Figure 1). Blue wells were counted to yield an estimate for the most probable number (MPN), an estimate of the CFUs per 100 milliliters present in the original water sample, for all detectable coliform bacteria in the American River. Using an MPN chart that came with the Coliplate™ microplate, the total number of blue wells, 60, (Figure 1) was match to its appropriate MPN of 255, suggesting the original water sample had 2.55 CFUs of coliforms per milliliter (Table 1). Likewise, the number of blue wells fluorescent under ultraviolet light indicated detection of E. coli in the tested river water sample. Only 4 wells were blue and fluorescent under UV light (Figure 1), which suggests that the E. coli content of the river was about 0.11 CFUs per milliliter (Table 1). Due to complications regarding proper storage of this microplate, there is a great likelihood that the obtained MPN values for total coliform and E. coli content in the river are overestimates, because the microplates were stored too long before being examined due to time management constraints.
| Characteristics of Wells | Number of Wells | MPN (Estimate of CFUs per 100 mL) | CFUs per milliliter |
|---|---|---|---|
| Blue color of any intensity under normal light | 60 | 255 CFUs per 100 mL | 2.55 CFU/mL |
| Blue color and fluorescent under UV light | 4 | 11 CFUs per 100 mL | 0.11 CFUs/mL |
Table 1: Observed blue well count and fluorescent blue well count for the river water sample.

Figure 1: ColiplateTM Microplate results: Wells under normal light on white background (left) and wells under ultraviolet light on black background (right).
To observe the total concentration of agar-friendly microbes in soil samples from the American River, different estimates of original cell density (OCD) were obtained for different dilutions of the River Water cultured on nutrient agar plates (Table 2). To yield an estimate of OCD, colony counts were controlled for volume of soil solution added and how diluted the solution was. On each plate, microbial colonies of different colors and shapes were observable (Figure 2). There was substantial variance among the three obtained estimates for OCD of the river water, and the quantity obtained for the OCD estimate increased by a factor of about 2.5 and 3 from the original dilution to the 1/10-dilution and from the 1/10 dilution to the 1/00 dilution respectively (Table 2). Regardless, use of the most conservative estimate of the original cell density for the American River suggests that a single milliliter of river water contains at least about 900 agar-culturable microbes.
| Selected Dilution of Soil Water | Number of CFUs in Agar Nutrient Plates | Estimated Original Cell Density (CFU/mL) |
|---|---|---|
| Original | 92 | 920 |
| 01:10 | 21 | 2100 |
| 1:100 | 6 | 6000 |
| Mean | N/A | 2659 |
| Standard Deviation | N/A | 3007 |
Table 2: The number of CFUs cultured in the agar nutrient plates for each soil solution and their derived estimates of original cell density of river bed soil solution.

Figure 2: Observed Colony Forming Units (CFUs) in the 100 (left), 10-1 (middle), and 10-2 (right) nutrient agar plates.
Corbicula fluminea feeding rates for E. coli and algae respectively were obtained by observation of change in microbe concentration over time. Different water samples were collected and measured for absorbance of light, a value directly proportional to the concentration of the respective microbe in the water sample; absorbance could not be used to obtain concentration since no molar extinction coefficient was available For the respective microbe, absorbance was plotted as a function of time on a scatterplot to yield a least squares regression line (LSRL) predicting sample absorbance as a function of time (Table 3). The negative slope for the LSRL was thus used as an estimate for the clam feeding rate, because microbial content in the aqueous environment surrounding the clam was expected to decrease substantially over time. E. coli and algal filter feeding rates were obtainable (Table 2) using the scatterplots for E. coli filtration (Figure 3) and algal filtration respectively (Figure 4). Each scatterplot yielded an R-squared value no greater than 0.0100, suggesting that no more than 1% of variance in sample absorbance could be explained by the variable of time. Each LSRL yielded a slopes of magnitude 10-5 absorbance per minute, almost negligible changes in absorbance per unit time. A negative filter feeding rate for algae feeding suggests that the quantity of algae in the environment may have increased at a small rate throughout the duration of the algal feeding experiment.
| Selected Microbe | Average Feeding Rate |
|---|---|
| Escherichia coli | +.00004 min-1 |
| Algae | -.00003 min-1 |
Table 3: Observed filter feeding rates.

Figure 3: Algae filter feeding by Asiatic clam.

Figure 4: E. coli filter feeding by Asiatic clam.
Six gill tissue specimens were extracted from different clams to inspect for spacing between their rows of cirri (Table 4). A boxplot was generated to display the spread of cirri spacing for all imaged gill tissues (Figure 5). The median mean cirri spacing for the six clams was 31 μm while the grand mean for cirri spacing among the six clams was observed to be 36 μm. In general, cirri spacing for clams was observed to be around 31 μm or 36 μm. C. fluminea cirri spacing observed in this lab was compared to literature cirri spacing for Pacific oysters (Crassostrea gigas), bivalves of comparable size to C. fluminea. It was thus observed that Asiatic clams have cirri spacing about twice as wide as that of Pacific oysters. A sample image of gill tissue explicitly showing cirri is provided in (Figure 6).
| Average Cirri Spacing | |
|---|---|
| Clam # 1 | 0.032 mm |
| Clam # 2 | 0.027 mm |
| Clam # 3 | 0.045 mm |
| Clam # 4 | 0.030 mm |
| Clam # 5 | 0.046 mm |
| Clam # 6 | 0.030 mm |
| Grand Mean | 0.036 mm |
| Standard Deviation | 0.0113 mm |
Table 4: Cirri spacing for observed Asiatic lams.

Figure 5: Spread of cirri spacing among observed Corbicula fluminea (n=6).

Figure 6: Example of Corbicula fluminea gill tissue specimen depicting eulateral frontal cirri (EFC).
DNA was extracted from three preserved C. fluminea tissue specimens to be amplified by PCR (Table 5). Only two of these amplified DNA extracts were used for agarose gel electrophoresis and DNA sequencing. The DNA sequence of interest was a ~700-base pair segment within the mitochondrial COI gene. Because the NEB 100-bp ladder was observable on the obtained gel image (Figure 7b), a semi log plot could be generated from the 1512-bp, 1200-bp, 1000-bp, 900-bp, 800-bp, and 700-bp bands. The semi log plot was thus used to predict DNA segment size (in base pairs) of a visible DNA band in another lane. Based on obtained semi log plot data, each lone band obtained for the C. fluminea mantle tissue samples were predicted to be about 750 base pairs long (Table 6). However, observation of the gel image suggests that the clam DNA bands have segments about 700-bp long, because the 700-bp band for the DNA ladder in the first lane is directly adjacent to the clam DNA bands. This observation helps reaffirm that the amplified DNA sequence was the COI mitochondrial gene. The C. elegans (roundworm) DNA used as a positive control was predicted to have a size of about 950 base pairs. A band with the same DNA segment length was detected in the lane for the negative control. This suggests that the negative control sample, which was not intended to contain DNA, was most likely contaminated with DNA from either of the DNA samples.
| C. fluminea | C. gigas | |
|---|---|---|
| Mean Cirri Spacing | 0.035 mm | 0.018 mm |
Table 5: Cirri spacing in Corbicula fluminea (Observed)vs. Crassostrea gigas (Literature)3.
| Sample | Distance from well (pixels) | Expected Sequence Length (bp) | Estimated Sequence Length (bp) |
|---|---|---|---|
| F18-0305-B1 (C. fluminea) | 171 | 700 bp | 750 bp |
| F18-0305-B3 (C. fluminea) | 171 | 700 bp | 750 bp |
| Positive Control (C. elegans) | 122 | N/A | 950 bp |
| Negative Control | 171 | 0 | 750 bp |
Table 6: Estimations of DNA sequence size from semi log plot.

Figure 7: Imaging of agarose gel electrophoresis of C. fluminea cytochrome C oxidase subunit I gene (labeled).

Figure 8: Semilog plot of agarose gel electrophoresis.
For the purposes of observing the COI gene in C. fluminea from the American River, two samples of PCR-amplified DNA from a single Asiatic clam was sent to another laboratory to sequence DNA using only a forward primer or a reverse primer respectively. An electropherogram and a computer-assigned nucleotide sequence were returned as a single file computer file for each sequenced DNA sample. Upon inspection of the electropherograms, it was observed that there is extensive overlap between colored plots for different nucleotides. To detect the nucleotide at a specific position in a DNA sequence, there should be only one peak on the electropherogram that corresponds to the nucleotide’s specific light wavelength. For both electropherograms, overwhelming proportion of sections had at least two or more overlapping peaks like the electropherogram section depicted in Figure 9. While the evidence is not definitive, the agarose gel electrophoresis data support the assertion that the sequenced DNA was from the COI gene of an Asiatic clam specimen (F180305B3). Even after trimming and substitution of assigned “N” nucleotides with more specific IUAC nucleic acid codes, there was no significant match to the sequence found in the NCBI database while conducting a “Nucleotide BLAST” search.

Figure 9: Representative section of poor DNA sequencing of C. fluminea cytochrome C oxidase subunit I gene using either forward or reverse primers.
Western blotting techniques were used to inspect C. fluminea gill tissue for any detectable trace of P-glycoprotein. SDS-PAGE was conducted to separate cellular proteins by size before transferring such proteins to PVDF membrane. The PVDF membrane was then subjected to application of primary antibody, successive washing, application of secondary antibody. Two substrates were added to the membrane for an enzyme covalently bound to the secondary antibody to yield fluorescent product. This fluorescent product was produced only at sites where secondary antibody annealed, yielding fluorescent bands at sites of P-gp or sites of indiscriminate binding. The bands formed are observable in Figure 10. It should be noted that the 100 kDa protein ladder was not detectable on the PVDF membrane image and thus a semi log plot could not be generated to verify the size of extracted P-gp. Significant fluorescent bands formed in the lanes containing positive control, proteins from gill tissue sample “1” (F180305A1), proteins from gill sample “2” (F180305A2), SU18R-NLA (200 ppm CaCl2), and SU18R-SUA2 (100 ppm CaCl2) (Figure 10). No bands were detectable in the lane for the 100 kDa DNA ladder or the lane of the negative control. Although fluorescence was observed for the CaCl2 samples, they will be ignored as they were not particularly relevant to this study.

Figure 10: Western Blot Results for 150kDa Protein (and CaCl2) Bands after Application of P-gp-specific Secondary Antibody and Chemiluminescent Substrate.
Because no band for the protein ladder formed, the widest most fluorescent band in the lane of the positive control sample (Figure 10) was presumed to be 150-kDa, the size of P-glycoprotein. Bands at the same distance from the well in each of the gill tissue protein extract sample lanes were also assumed to be 150-kDa and thus P-gp. The chemiluminescence of each lane at the 150-kDa section was measured and compared as measure of relative P-gp content, as depicted in the bar chart for Figure 11. The positive control was observed to have the most fluorescent band, which was assigned a value of 24670.853, (Figure 11) while gill tissue sample “1” (F180305A1) and gill sample “2” (F180305A2) were assigned values of 2040.619 and 7291.61 respectively. This suggests that the first gill tissue had about 8.3% the P-gp content of the positive control while the second gill tissue sample had about 29.5% of the P-gp content of the positive control.

Figure 11: Chemiluminescence of Protein (and CaCl2) Bands after Application of Secondary Antibody and Substrate.
Discussion
Due to issues of access to experimental resources, the Coliplate™ microplates used in this experiment could not be viewed for one week after they began incubation. The well count-dependent MPN values provided are based on incubation of the microplates after twentyfour hours. In hopes of suppressing additional microbial growth, the microplates were placed into a freezer colder than 0°C for 122 hours by laboratory technicians after incubation of the microplate for about 24 hours. While microbial growth may have been inhibited substantially, the MPN estimates of the E. coli and total coliform concentrations in the American River are likely to be overestimates of the true microbial concentrations in the river. Despite these issues of overestimation, the collected evidence that E. coli and other coliforms are present in the American River. The collected data suggest that 4.3% (11/255) of such coliforms in the American River are E. coli. Because there was a great number of blue-colored wells due to the extended incubation time, it could be argued that the great incubation time provided a greater sample size (n=60) to determine relative abundancy of E. coli with respect to all detectable coliforms in the American River. Future studies using Coliplate™ microplates or related techniques to detect E. coli and total coliform concentrations could draw from this study if intent is to compare the relative frequency of E. coli with respect to all detectable coliforms in an aqueous sample.
Fewer inferences can be drawn from the effort to determine the original cell density of the soil solution. The standard deviation of these three estimates was observed to be larger than the mean OCD estimate, suggesting that the sample size (n=3) used was far too small. Better estimates of river water OCD could be obtained if the more river water samples were diluted and cultured to yield a better estimate and smaller standard deviation. The validity of original cell densitydilution technique for estimating microbial contents of water could also be tested by means of a one-by-three way ANOVA F-test. While the only one river sample was diluted in this procedure, it is alarming that such a technique yielded such a great spread in estimated microbial concentrations (OCD values).
Flaws in experimental design likely contributed to the lack of significant observable E. coli algal feeding rates in the tested Asiatic clam. The same clam was used in both filter-feeding trials to control for many potential confounding variables. If more resources were available, multiple clams could have been subjected to a single condition so there could be statistical analysis of two treatment groups. For the observed relationships between absorbance of an aqueous sample and time, it should be noted that R-squared values of less than 0.0100 suggest that less than 1% of variation among the absorbance of aqueous samples in this study could be explained by time. This strongly suggests that the studied clam was not filter feeding too much at all. There were multiple instances where the clam was observed to extend its siphon for extended durations near the beginning of the algal feeding experiment. After the first forty-five minutes, the clam was observed to have its shells closed together tightly. Throughout the duration of the second filter feeding experiment using E. coli, the clam was only observed to maintain an adducted position, never to open its siphon. This lack of behavior likely explains the low estimated filter feeding rates based on collected data. The lack of such information would contradict studies that suggest C. fluminea is a great filter feeder. It has been reported that C. fluminea filter feed at rates three times that of the zebra mussel (Dreissena polymorpha) after controlling for surface area of filtering tissues. Asiatic clams are also reported to filter 50-μm algal particles and particles as small as1 μm, suggesting that particle size is not an issue. Future studies measuring filtration rates in bivalves are advised to conduct multiple trials with multiple clams to more effectively control for confounding variables.
The spacing between rows of cirri was measured in six different Asiatic clams. The grand mean for the spacing between different adjacent rows of cirri was observed to be 36 μm with a standard deviation of about 1.1 μm. The median for mean cirri spacing among the six clams was 31 μm. These data suggest that the cirri spacing in mature Asian clams may generally be between 31 μm and 36 μm. C. fluminea observed in this studied were observed to have spacing almost twice as much as the 18-μm spacing in Pacific oysters (Crassostrea gigas), a bivalve of comparable size. While the direct implications of our spacing findings are limited, further research that investigates a relationship between cirri spacing and filtration rates may be of value. C. fluminea has a great biofiltration capacity relative to other bivalves of similar size, and it may thus be valuable to investigate whether cirri spacing is among the most important of variables contributing to filtration abilities.
Even after trimming and other manipulation of the assigned DNA sequence, the electropherogram results for the amplified cytochrome C oxidase subunit I (COI) gene was practically unsalvageable for both the forward and reverse primer-sequenced mtDNA samples. While no definitive evidence could prove that the COI gene was the gene sequenced in the other lab, the agarose gel electrophoresis results suggest that amplified gene had a size estimated to be any size between 700 bp and 750 bp, close to the reported ~700-bp size of the COI gene generally used for DNA barcoding of animals [10]. The consistency of the size of the DNA segments spread on agarose gel with literature values rules out issues in sequencing data due to improper cell lysis and DNA amplification techniques. The primers used for sequencing were also shown by “Nucleotide BLAST” search results perfectly matched the COI gene of animals in entirely different phyla than bivalves, like mosquitos, crustaceans, nematodes, and ribbon worms [12].
The lack of a visible protein ladder in the PVDF membrane imaging did not inhibit our findings too greatly. While it was not possible to check for the expected protein size using a semi log plot based on ladder data, the formation of thick, highly chemiluminescent band for the positive control sample was sufficient enough to determine that adjacent bands were likely P-glycoprotein (P-gp), a 150-kDa ABC transporter protein attributed to multixenobiotic resistance in bivalves [7]. The chemiluminescence of each band could not be properly attributed units due to limitations of software, but the relative chemiluminescence of each gill tissue-sourced protein band could be compared to the positive control. One gill tissue was observed to have a protein band that was about 3.5 times more chemiluminescent than the band of the adjacent gill tissue sample, suggesting a substantial difference in P-gp contents between the gill tissues of the source C. fluminea specimens. The detected presence of P-gp in these two Asiatic clams may support the assertion that C. fluminea exhibit multixenobiotic resistance like other bivalves such as the eastern oyster (Crassostrea virginica). Before making the assertion the studied clam with greater P-gp content may have been exposed to a greater concentration of xenobiotics, it is worth mentioning that P-gp content is not observed to be directly related to P-gp activity.6 Different studies have observed only a 100-150% increase in P-gp activity in response to environmental conditions that effectively increased P-gp levels by 250-600%. Due to their similarities in composition, mitochondrial P-gp—whose functions are not currently understood6—may have been detected in addition to the cell membrane P-gp of interest that has specifically been attributed to multixenobiotic resistance.
Conclusion
This study was conducted to better understand the influence of the invasive species Corbicula fluminea on the American River, a freshwater ecosystem in the Sacramento region, and the river’s microbial contents. To better understand the mechanisms that yield such efficient and broad filtration abilities, C. fluminea were tested for their filtration abilities when subjected to different microbes, but the observed feeding rates for E. coli and algae were close to zero. Use of original cell density calculations to estimate agar-cultural microbe concentration in the river bed soil yielded too large of a spread to provide a precise estimate. Estimations yielded for E. coli content and total coliform content in water of the American River were overestimates due to procedural limitations. However, a rough estimate of 4.3% was obtained to describe the relative abundance of E. coli with respect to all detectable coliforms in the American River. Based on gill tissue extracts from six clams, it was determined that the cirri spacing in C. fluminea was around 31 to 35 μm, a large length for bivalves of comparable size. Amplified extract of the C. fluminea cytochrome C oxidase subunit I (COI) gene was run on agarose gel and sequenced, but the two yielded electropherograms were so poor that trimming of the accompanying sequence could not make it salvageable. P-glycoprotein was detectable in two different C. fluminea gill tissue specimens in different relative abundance where one clam was observed to have about 3.5 as much P-gp as the other. While P-gp traces were detectable, resources were limited for yielding proper concentration values for the P-gp detection and filter feeding procedures. Further research is needed to better understand the influence of cirri spacing on filtration rates in bivalves. Additional investigation into the influence of P-gp on multixenobiotic resistance mechanisms may also be useful to better quantify the multixenobiotic resistance exhibited by bivalves of different species.
References
- Gomes J, Matos A, Quinta-Ferriera, R M Martins, R C (2018) Environmental Applications of Invasive Bivalves for Water and Wastewater Decontamination. Sci Total Environ 630: 1016-2017.
- https://www.mayoclinic.org/diseases-conditions/e-coli/symptoms-causes/syc-20372058
- Agbaje OBA, Shir I B, Zax DB, Schmidt A (2018) Biomacromolecules within Bivalve Shells: Is Chitin Abundant? Acta Biomaterialia 80:176-186.
- Spann N, Harper E M, Aldridge D C (2010) The Unusual Mineral Vaterite in Shells of Freshwater Bivalve Corbicula fluminea from the UK. Naturwissenschaften 97:743-751.
- Arryl L, Cain M L, Wasserman, SA, Minorsky PV, Reece JB (2017) An Introduction to Invertebrates. Campbell Biology 11 Pearson: New York 697-700.
- Ivanina AV, Sokolova I M (2008) Effects of Cadmium Exposure on Expression of P-glycoprotein in Eastern Oysters Crassostrea virginica Gmelin. Aqauat. Toxicol 88: 19-28.
- Guo X, Feng C (2018) Biological Toxicity Response of Asian Clam (Corbicula fluminea) to Pollutants in Surface Water and Sediment. Sci Total Environ631:56-70.
- Kurelec B, Waldmann P, Zahn R K (1996) The Modulation of Protective Effects of the Multixenobiotic Resistance Mechanism in a Clam Corbicula fluminea. Mar Environ46: 383-387.
- Zaja R., Klobucar GI.V, Klobucar R. S , Hackenberger BK, Smital T (2006) Haemolymph as Compartment for Efficient and Non-destructive Determination of P-glycoprotein (Pgp) Mediated MXR Activity in Bivalves. Compa Biochem Physiol Part C 143: 103-112.
- Kress W J, Garcia-Robledo C, Uriarte M, Erickson D L (2015) DNA Barcodes for Ecology, Evolution, and Conservation. Trends Ecol Evol.30: 25-35.
- Kowalska Z, Pniewski F, Latala A (2019) DNA Barcoding ‒ A New Device in Phycologist’s Toolbox. Ecohydrol Hydrobiol.
- Barille L, Haure J, Cognie B, Leroy A (2000) Variations in Pallial Organs and Eulatero-frontal Cirri in Response to High Particulate Matter in Concentrations of the Oyster Crassostrea gigas. Can J Fisheries and Aquatic Sciences 57, 837-843.
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Indexed at, Google Scholar, Crossref
Citation: Talebzadeh A (2023) Investigation of Invasive Species Corbicula Fluminea and Microbial Contents of the American River. J Ecosys Ecograph 13: 390. DOI: 10.4172/2157-7625.1000390
Copyright: © 2023 Talebzadeh A. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 2223
- [From(publication date): 0-2023 - Sep 03, 2025]
- Breakdown by view type
- HTML page views: 1914
- PDF downloads: 309