The Receptor for Advanced Glycation Endproducts (RAGE) and Mediation of Inflammatory Neurodegeneration
Received: 06-Jan-2018 / Accepted Date: 17-Jan-2018 / Published Date: 24-Jan-2018 DOI: 10.4172/2161-0460.1000421
Abstract
The Receptor for Advanced Glycation Endproducts (RAGE) is an immunoglobulin-type, transmembrane receptor that is expressed on numerous cell types in the Central Nervous System (CNS) and periphery, such as neurons, astrocytes, microglia, mononuclear phagocytes, epithelial cells and endothelial cells (ECs). RAGE binds a discrete repertoire of ligands, including non-enzymatically glycated proteins and lipids, also known as advanced glycation endproducts (AGEs), for which the receptor is named, in addition to multiple members of the S100/calgranulin family, oligomeric forms of Aβ, high mobility group box 1 (HMGB1), phosphatidylserine (PS) and lysophosphatidic acid.
Keywords: Alzheimer’s disease (AD); Cerebrovascular ischemia (CI); Parkinson’s disease (PD); Amyotrophic lateral sclerosis (ALS); Dementia
Introduction
Extensive evidence has implicated RAGE as a critical player in regulating inflammation, as well as oxidative and cellular stress in a variety of organ niches and disease settings, including the CNS during neurodegeneration [1-14].
This review will focus on the current state of knowledge regarding RAGE and neurodegeneration. Specifically, we will detail the effect of RAGE signal transduction on cellular stress, pinpoint clues into RAGE pathophysiology in the context(s) of increased RAGE ligand burden, discuss the systemic consequences of RAGE-driven inflammation in the CNS as a whole, and report on the increasing number of published genome wide association study (GWAS) findings and studies reporting on biomarkers of RAGE activity that collectively evoke strong indications for RAGE as a putative driver of cellular and systemic dysfunction during key neurodegenerative pathologies, most specifically Alzheimer’s disease (AD), ischemic cerebrovascular disease, Parkinson’s disease (PD), Amyotrophic Lateral Sclerosis (ALS), frontotemporal dementia (FTD) and Multiple Sclerosis (MS).
Consequences Of RAGE Signal Transduction
Our laboratory recently discovered that upon ligand engagement of the extracellular domains of RAGE, the RAGE cytoplasmic domain binds to its intracellular effector molecule, Diaphanous 1 (DIAPH1) [15,16]. DIAPH1 has subsequently been shown to be required for signal transduction induced by RAGE ligand binding, including the activation of mitogen activated protein kinases (MAPK), Rho GTPases and phosphatidylinositol 3-kinase (PI3K)/Akt signaling. RAGEDIAPH1 signaling effects are dependent on many factors, including, but not limited to: cell-type, ligand form and ligand concentration, and the duration of signal induction (acute vs. chronic) [17-22].The implications of activation of these signaling cascades are substantial and predominantly pathological. The RAGE-DIAPH1 interaction drives the generation of reactive oxygen species (ROS), the induction of cellular migration, the upregulation of inflammatory cytokines and subsequent downregulation of ATP binding cassette (ABC) cholesterol transporters, such as ABCA1 and ABCG1, thereby mediating intracellular lipid accumulation and consequent cellular dysfunction [9,23-25].
RAGE signaling can directly impact mitochondrial health and function by modulating mitochondrial fission, ATP production, membrane potential and by promoting mitochondrial death pathways [26-30]. In pathological environments, RAGE signaling-induced cytosolic ROS production can promote production of mitochondrial ROS, thereby amplifying total ROS production [31-33].
Besides its role in RAGE-DIAPH1-mediated inflammation, DIAPH1 is a dynamic mediator of actin cytoskeleton stability and rearrangement, as well as a regulator of transcription factors [15,34-36]. It was recently reported that DIAPH1 was highly expressed in human gliomas; however, the specific details of DIAPH1 expression, including the cellular localization and the potential DIAPH1-mediated mechanisms of in vivo dysfunction in the rodent or human CNS, have not been elucidated [37]. Beyond this report, very little is known about DIAPH1 expression patterns and functions in the CNS of normal or degenerating models of humans; there are no known SNPs in DIAPH1 that increase or decrease neurodegenerative disease risk. However, the impact of RAGE-DIAPH1 signal transduction in peripheral cells exhibits prominent overlap with the patterns of cellular dysfunction observed in neurodegeneration, including the increased production of ROS and pro-inflammatory cytokines and the downregulation of homeostatic molecules, such as neurotrophins and cholesterol/lipid handlers. This signaling culminates in significant alterations in critical cellular functions, such as migration, phagocytosis, replication and cell death, particularly in cells of myeloid and endothelial origin, but also in neurons [4,12,38-40].
Connecting the Dots: Potential RAGE Mechanisms in Alzheimer's Disease
Alzheimer’s disease (AD) is a neurodegenerative disorder that impacts millions of people worldwide and is not curable. While the primary risk factor for AD is advanced age, recent insights from genomic technology implicate inflammatory lipid and cytokine signaling in microglia, the myeloid cells of the CNS, as a prominent correlate of disease. Specifically, human GWAS suggest a powerful link between inflammatory pathways, including complement, chemokines and influential lipid and cholesterol molecules, such as Triggering Receptor Expressed on Myeloid Cells 2 (TREM2), ABCA7, Apolipoprotein E variant 4 (APOE4) and others with AD susceptibility [14,20,41-49]. Additional analyses within animal models have illuminated various molecules critical to the innate immune system as major contributors to increased or decreased rate of AD progression, such as Chemokine Receptor Type 2 (CCR2), Chemokine Receptor 1 (CX3CR1 or GPR1), complement components (C1q and C3) and Chemokine Ligand 8 (CXCL8) [43,50-63].
The most prominent risk alleles and impairments were observed in humans and mice with loss-of-function mutations or deletions of the aforementioned chief lipid handling molecules. However, burgeoning data in humans and rodent models also indicate that systemic inflammation and transient infections in the periphery are sufficient to increase production of RAGE ligands, particularly AGEs and oligomeric Aβ. In contexts in which these ligands accumulate in the CNS, RAGE signaling is causally implicated in exacerbating ongoing neurodegenerative disease. In addition, RAGE has been shown to mediate mitochondrial dysfunction in neurons by transporting Aβ into the cells, which subsequently results in greater neuronal dysfunction and degeneration [64]. Atop the multiple mechanisms of augmented RAGE ligand production in AD, there is also prominent downregulation of specific detoxification mechanisms, which inhibit production of pre-AGEs such as methylglyoxal (MG) [65]. Glyoxalase 1 (GLO1), the principal enzyme that detoxifies MG, mitigates AGE production and is upregulated in the early and mid-stages of AD in human subjects. However, in the late and progressive stages of AD dysfunction, depletion of the enzyme’s chief and essential cofactor, glutathione, reduces overall activity of the GLO1-AGE detoxifying system, thus facilitating increased AGE production and accumulation [65,66]. Altogether, these findings underscore a potentially profound link between inflammation, both peripheral and central, which prompts the question: to what extent might anti-AGE/RAGE therapies provide protective measures for neurodegeneration and AD, given the prominence of cellular stress driven by increased RAGE ligand burden [67-69].
Population-based studies have emerged suggesting links between RAGE, dementia and AD. Genetic sequence variations in 20 genes associated with inflammatory signaling were recently probed for possible associations with dementia risk. From 1,462 Swedish dementia cases and 1,929 controls that were composed of twin and unrelated case-control samples, investigators identified a potential association of sequence variations near the gene encoding RAGE (AGER), to increased risk for dementia and AD, in two independent samples. Further, a recent structural analysis utilizing MRI technology revealed that atrophy of the right hippocampus substructure CA1 during AD progression was significantly correlated to the single nucleotide polymorphism (SNP) variant rs2070600 within AGER [70]. Notably, this variant has been previously associated with increased affinity to ligands and increased ligand-stimulated inflammation in cultured cells, in conjunction with decreased levels of circulating soluble RAGE (sRAGE) [71]. sRAGE is a short, soluble isoform of RAGE, and putative “decoy” receptor. Because it lacks the intracellular and cytoplasmic domains required for signaling, sRAGE is predicted to protect against inflammation and RAGE-dependent cellular stress by sequestering RAGE ligands and preventing their engagement of the full-length, transmembrane RAGE [72,73]. Thus, in humans bearing this SNP, lower sRAGE concentrations may directly amplify ligand burden and availability for signal transduction through full-length RAGE. This increases cellular stress, impairs lipid and cholesterol handling for the cells, in addition to promoting increased ROS production, thereby forging a feed-forward, self-perpetuating loop of inflammatory cellular stress in ECs, myeloid cells, and others within the CNS niche, including astrocytes, neurons, and oligodendrocytes.
Many of the mechanistic studies of RAGE in AD-like mouse models have been conducted in animals that are globally devoid of Ager and animals with dominant negative-RAGE (DN-RAGE) targeted to myeloid cells, using the macrophage scavenger receptor promoter. DN-RAGE is composed of the extracellular RAGE domains and the transmembrane domain; hence, although ligand binding to this construct is intact and it is tethered to the cell membrane, signaling is abrogated on account of deletion of the cytoplasmic domain. These DN-RAGE studies have indicated that RAGE signal abrogation confers a benefit for AD progression and suggest a role for RAGE in myeloid cells during AD [10,12,47,74]. However, there are possible caveats to these studies, particularly since it is plausible that DN-RAGE may also act as a decoy receptor and “ligand sink”, much like sRAGE, and mice devoid of Ager or expressing DN-RAGE constitutively from birth may develop differently than a wild-type animal. Therefore, further investigation utilizing greater cell type- and temporal specificity would be key for definitively determining a role for RAGE in AD.
RAGE molecules expressed on ECs are also known to facilitate the transport of Aβ into and across the blood brain barrier (BBB) during AD, implicating RAGE in mediating the increased pools of ligand concentrations found during disease progression [9,75]. Since AGE production is increased in oxidized environments and RAGE engagement drives ROS production, there are additional entry points into the aforementioned feed-forward loop in which RAGE ligand binding drives increased RAGE ligand abundance, increased RAGEDIAPH1 signaling and therefore increased ROS and AGEs. Together, this AGE-generating loop and the reduced expression of Low Density Lipoprotein Receptor-related Protein 1 (LRP1), the chief molecule responsible for transporting Aβ out of the brain in AD, collectively dysregulate the flux and trapping of AGEs and Aβ within the CNS as degeneration progresses [76]. Collectively, these data provide strong evidence for the RAGE-DIAPH1 signaling axis as a prominent mediator of inflammation and cellular dysfunction in a variety of cell types during AD, particularly by igniting an unconstrained iterative loop of signal propagation driving cell-intrinsic and cell-to-cell stress signals that mediate prominent impairments during AD.
Of note, the extracellular RAGE inhibitor, Azeliragon, is currently in Stage 3 clinical trials to investigate the therapeutic potential of RAGE inhibition in AD patients. Initially, in an 18 month Stage 2 clinical trial of 399 patients, the trial was preemptively halted when Azeliragon (then by the name of TPP488) was shown to be deleterious to patients at high doses (60 mg for 6 days followed by 20 mg), but protective at low doses (15 mg for 6 days followed by 5 mg) [77,78]. Currently, a Stage 3 study granted Fast Track designation by the United States Food and Drug Administration is being conducted that utilizes the low dose (5 mg for 18 months) vs. placebo. This trial, entitled the STEADFAST Study, was recently extended for an optional 2 year continuation in multiple countries across the world [79].
RAGE and Ischemic Cerebrovascular Disease: Acute and Chronic Implications
Acute and chronic ischemia of the brain leads to dramatic alterations in the health of the CNS, regardless of the mode of impact. Whether induced by stroke, cardiovascular disease, traumatic brain injury or pharmacological models of human disease, a large body of work has consistently linked cerebral ischemia to increased expression of RAGE and its ligands, particularly HMGB1, in the affected brain tissue. The same AGER SNP, rs2070600, associated with increased risk ratios for the development and progression of AD, has also been shown to be associated with increased risk of ischemic stroke (and Coronary Artery Disease), particularly in Chinese populations [80]. In addition, in assessments of specific sRAGE subtypes, increased levels of cell surface-cleaved soluble RAGE81]. Beyond this, very little is known about RAGE and its ligands in human manifestations of cerebral ischemia, although mechanistic studies in rodents may provide further lines of evidence for better understanding the ways in which RAGE contributes to the devastating effects of ischemic cerebrovascular disease.
Many studies in various rodent models utilize in vivo and ex vivo models of transient ischemia to study the impact of stroke and downstream adverse events of this acute event, including central post-stroke pain (CPSP) and connections to increased risk of AD. In particular, the transient middle cerebral artery occlusion (tMCAo) stroke model has been invaluable in illuminating potential roles for RAGE signaling in ischemia. Upon induction of ischemia by tMCAo, mice show an immediate and robust increase of RAGE expression in the striatum and cortex; however, inhibition of nitric oxide synthase in ECs in these regions greatly exacerbated these effects and led to an increased expression of IL6, TNFα and RAGE [82]. Beyond receptor upregulation, subsequent mass spectroscopy studies have demonstrated that HMGB1 is upregulated in the cerebrum, spinal cord, and carotid nerve of mice and rats after ischemia [83,84]. Peripheral HMGB1 upregulation has been shown to be a specific driver of “sickness behavior” in the hyperacute injury recovery period and neutralization of HMGB1 and/or cytokines was shown to be protective for these behaviors and able to diminish peripheral immune exhaustion, which has been frequently observed after cerebral ischemia [83]. This work has paved the way for some of the most profound studies connecting RAGE regulation of the peripheral immune system and specific impacts on CNS health and disease-related behavioral abnormalities, perhaps most strikingly because cerebral ischemia, by definition, involves breakdown of the BBB and the consequent infiltration of peripheral RAGE-positive monocytes.
Subsequent work has also highlighted profound implications for hyperglycemia driving increased infarct volume and a decreased number of protective, non-inflammatory monocytes and macrophages infiltrating the injured CNS brain regions. Specifically, these studies have shown that the ablation of peripheral monocytes or RAGE/HMGB1 inhibition in peripheral monocytes, through genetic ablation of Ager or Hmgb1, provides benefits to mice with respect to hyperglycemiainduced impairments in stroke rehabilitation, including: decreased infarct area, prevention of BBB leakage, and decreased HMGB1 and RAGE expression, specifically in the cerebrum and microglia [85,86]. These findings were further supported by a subsequent study utilizing WT and AD-like mouse hippocampal slices subjected to oxygen glucose deprivation in the presence or absence of synthetic Aβ oligomers and showed that DN-RAGE targeted to the macrophage scavenger receptor promoter protected animals from ischemia and/or Aβ-induced synaptic impairments, thus implicating microglia RAGE in driving further detriments in ischemic cerebrovascular disease [87]. However, the previously mentioned caveats of DN-RAGE still pertain, and further mechanistic study of these findings utilizing more precise models would be helpful in elucidating the roles of central vs. peripheral myeloid cells and how other CNS cells, particularly ECs, are involved in RAGE-dependent cerebral ischemic impairments.
RAGE and Parkinson's Disease, Another Manifestation of Cellular Dysfunction
Parkinson’s disease (PD) is another common neurodegenerative disorder that impacts millions of people worldwide and is characterized by the specific loss of nigrostriatal dopaminergic neurons and locomotor deficits [88]. While the cerebral location and neuronal subsets that degenerate in PD are distinct from AD, there are prominent cellular activation mechanisms driving inflammation and perturbation of neurons at the nexus of the two disorders. Akin to AD, the initiation of PD pathogenesis is still not clearly elucidated. However, there are many disease processes correlated to PD and AD pathogeneses, which could potentially be related to RAGE-DIAPH1 signaling, such as enhanced oxidative stress, innate immune activation, protein aggregation and neuronal death.
Multiple lines of evidence suggest a potential role for RAGE and its ligands in the pathogenesis of PD. First, the same AGER rs2070600 SNP that was implicated in CA1 atrophy during AD, was also correlated to the highest risk for PD development of all known AGER SNPs in a Turkish cohort GWAS (N=174 PD patients and N=150 healthy controls) [89]. In addition, when compared to healthy controls, PD patients have recently been shown to possess higher concentrations of RAGE ligands S100B and HMGB1 in the substantia nigra and cerebral spinal fluid (CSF) [90-92]. In rodent models, numerous studies have indicated that animals derive prominent protection from PD-like impairments when RAGE signaling was blocked through genetic ablation of S100B/RAGE or by the administration of a RAGE inhibitor, FPS-ZM1, a BBB permeable, high affinity, multimodal blocker of RAGE [90]. Either strategy was sufficient to abrogate a variety of impairments observed in the PD-like rodent models, such as apoptosis of dopaminergic cells; locomotor defects; neuroinflammatory microgliosis and astrogliosis, as measured by increased ionized calcium binding adaptor molecule 1 (IBA1) and glial fibrillary acidic protein (GFAP) staining, respectively; tyrosine hydroxylase (and therefore dopamine) deficits; NF-KB activation; and tumor necrosis factor alpha (TNFα) upregulation in the presence of PD-like syndromes induced by toxins. While many of these benefits only partially rescued cellular deficits or delayed the onset of disease, it is possible that RAGE-based interventions in AD and PD may provide meaningful avenues for therapeutic intervention in either condition of neurodegeneration.
Amyotrophic Lateral Sclerosis, Another Inflammatory Syndrome of the CNS?
Amyotrophic Lateral Sclerosis (ALS) is a fatal neurodegenerative disorder characterized by progressive loss of motor function and muscle atrophy. Much like AD and PD, many of the gene mutations linked to ALS have also been shown to drive inflammatory glial activation, oxidative stress, and neuronal loss. There is prominent overlap of disease phenotypes in ALS to other disorders with regard to the cellular consequences of RAGE-DIAPH1 signaling, although further investigation is required to elucidate these mechanisms [93]. Several studies have reported increased concentrations of RAGE ligands in the spinal cord [94-96] and CSF of ALS patients [97]. Conversely, serum sRAGE was decreased in human ALS patients, thereby putatively increasing ligand burden available for binding to and inducing signaling through full-length RAGE [98]. While little mechanistic evidence is available linking RAGE and ALS, our laboratory recently showed that RAGE and its ligands are increased in the spinal cord of ALS patients [95].
Furthermore, this increase of RAGE and its ligands was recapitulated in one of the most commonly employed ALS rodent models, murine lines containing the familial G93A mutation in superoxide dismutase 1 (SOD1), as discovered in human ALS populations [3,99,100]. In these models, nerve growth factor (NGF) is post-translationally modified by oxidation and contributes to RAGE signaling-induced motor neuron death when normal motor neurons are co-cultured with SOD1 G93A astrocytes [101]. In addition, C6 rat astrocytoma cells overexpressing mutant SOD1 G93A protein displayed significantly increased RAGE ligand S100B expression and, intriguingly, inhibition of this process by siRNA targeting S100b ameliorated the inflammatory profile of these cells [3].
In the SOD1 G93A mice, daily administration of recombinant sRAGE extended lifespan and duration of healthy body weight, while slowing the onset of motor function loss [99]. Importantly, sRAGE treatment not only reduced motor neuron death but also decreased astrogliosis, indicating a more homeostatic profile in multiple cell types [99]. Altogether, a burgeoning body of literature suggests that RAGE activation, driven by an increased availability of ligands, is likely a contributing factor to ALS pathology. However, further work utilizing established BBB-permeable inhibitors of RAGE-DIAPH1 would be paramount in elucidating the value of targeting this signaling axis as a potential therapeutic target for slowing the progression inflammatory and neuron-perturbing signaling in ALS.
There is growing evidence that frontotemporal dementia (FTD) and ALS share certain molecular pathologies; in fact, a subset of ALS patients also exhibits behavior phenotypes of FTD [102]. Multiple genetic mutations have been linked to both ALS and FTD, including mutations within the genes: C9orf72, CHCD10, SQSTM1 and TBK1, which contribute to RNA metabolism, autophagy, mitochondrial health, and microglial function [102,103]. Two reports have provided evidence of increased RAGE ligands in the CSF and cortex of FTD patients relative to control patients [104,105]. Altogether, there is probable involvement of RAGE to FTD-associated pathology when considering the increased RAGE ligands and the emerging molecular pathology overlap with ALS. It will be important to study RAGE signaling in the context of ALS/FTD mouse models such as C9orf72 hexanucleotide repeat expansion transgenic mice to determine if approaches used to treat ALS would have any benefit to the cognitive pathologies associated with FTD.
Multiple Sclerosis And Experimental Autoimmune Encephalopathy (EAE)
Multiple Sclerosis (MS) is a debilitating neurodegenerative disease in which autoimmune tissue-destructive processes are implicated. In human subjects, the AGER rs2070600 SNP was associated with MS in several studies [72,106]. However, in a different study of a Hungarian community, this SNP was not identified. Although, another SNP within the AGER promoter suggested that altered transcription, rather than differences in ligand binding and sRAGE production, may be contributing to the risk of MS within this population [107].
With respect to sRAGE, akin to other inflammatory neurodegenerative syndromes discussed above, MS patients display lower serum levels of sRAGE relative to control patients and this decreased sRAGE inversely correlates with disease progression [108]. In addition, RAGE ligands are also increased in active MS lesions, as observed by immunohistochemistry. Further, AGER mRNA and RAGE ligand protein concentrations were increased in serum, CSF, and mononuclear cells in both niches during MS [108-112]. Interestingly, patients treated with disease-modifying drugs display a prominent reduction of serum HMGB1 when compared to untreated MS patients, which correlated to a better disease prognosis [111]. Fingolimod, a sphingosine-1P (S1P) analogue, has also been utilized to treat relapseremitting MS in human patients, and induces a significant reduction in serum HMGB1 after 6 months of treatment while increasing sRAGE, albeit this study was conducted in a small patient cohort (n=17) [112].
Induction of experimental autoimmune encephalomyelitis (EAE), in which mice are immunized with myelin basic protein (MBP), has been utilized to study the molecular mechanisms underlying MS. Studies have reported increased RAGE expression in the spinal cords of mice with EAE [109,113], whereas blockade of RAGE signaling by recombinant sRAGE administered concomitantly with EAE induction in mice, significantly reduced immune cell infiltration into the brain and the severity of the disease [113]. However, controversy arose after a report that Ager deficient mice with EAE displayed no differences in disease severity [67].
Three distinct, but not exclusive possibilities may explain these seemingly conflicting results. First, recombinant sRAGE may exert some of its effects independent of RAGE signaling. It is possible that sRAGE is functioning as a pathological ligand sink in this instance that not only reduces RAGE signaling but other inflammatory signals as well through different receptors to which RAGE ligands may also bind. Second, the deletion of Ager from every cell may imbue detrimental effects due to unknown roles of RAGE in homeostatic functions and thus, a complete blockade of this signaling, as opposed to dampening, may reduce the benefits of RAGE inhibition. Third, it is well established that MS and EAE models in mice are characterized by periods of exacerbation vs. remittance of disease; hence, the timing of RAGE inhibition or Ager deletion in vivo may critically impact phenotypic outcomes.
Collectively, these considerations suggest that RAGE signaling is likely contributing to inflammatory perturbation in MS. Potential therapeutic interventions should investigate the possibilities of abrogating disease pathology by quenching RAGE ligands and/or preventing RAGE inflammatory signaling as well, although a much more detailed analysis of when and how to do so would still need to be conducted.
Conclusion
As summarized in the Table 1 and Figure 1, the manifestations of AD, PD, ALS and MS are distinct in nature, impacting differential subsets of neurons and regional variability within the CNS. However, there are common underlying threads that strongly suggest similarities among these neurodegeneration syndromes, including increased accumulation of RAGE ligands and expression of RAGE, processes that trigger oxidative and cellular stress, and myeloid, neuronal, astrocytic and endothelial dysfunction. The last decade of research has generated a formidable body of evidence to suggest that RAGE signaling plays a prominent role in the pathophysiology of these inflammatory neurodegenerative syndromes, although many of the specific details remain to be fully elucidated. Although these findings are illuminative, multiple questions remain to be addressed, such as does RAGE signaling participates in disease induction and/or as a potentiation/progression mechanism in these disorders? Why do we sometimes discern differential outcomes upon the use of sRAGE, RAGE inhibitors, or, in animal models, introduction of DN-RAGE expression or genetic ablation of Ager? To what extent does RAGE play time-dependent roles during discrete periods of disease and in distinct cell types, in models vs. humans, and are the effects of RAGE specific to aging or prominent across the lifespan? Are there specific patient populations for which RAGE-based therapies would be most or least beneficial? To this end, the future application of recent insights from human GWAS data for the use of genetic testing in conjunction with measuring circulating sRAGE levels might be the first steps to determine the subpopulations in which the administration of RAGE inhibitors may increase healthspan. RAGE presents itself as an attractive target for inhibition, when aiming for therapies that assuage cognitive decline during neurodegeneration through interfering with feed-forward loops of inflammation and oxidative and cellular stress.
| RAGE SNPs | Animal Findings | Human Findings | |
|---|---|---|---|
| Alzheimer’s disease | rs2070600 | 1. Increased RAGE and ligands 2. Mitochondrial Dysfunction 3. Decreased detoxification molecules 4. Decreased sRAGE 5. Oxidative Stress 6. Microglia and ECs implicated |
1. Increased RAGE and ligands 2. Hippocampal atrophy associated with SNP 3. Decreased sRAGE |
| Ischemic Cerebrovascular disease | rs2070600 | 1. Increased RAGE and ligands 2. Exacerbated by hyperglycemia in RAGE-dependent manner 3. Strong connections to peripheral monocytes, microglia and ECs |
1. Increased RAGE and ligands 2. cRAGE increased acutely after stroke 3. RAGE+ Monocyte infiltration |
| Parkinson’s disease | rs2070600 | 1. Increased RAGE and ligands 2. RAGE exacerbates all known symptoms of disease in models |
1. Increased RAGE and ligands |
| Amyotrophic Lateral Sclerosis and FTD | No known SNPs associated | 1. Increased RAGE and ligands 2. RAGE-induced motor neuron death 3. Stronger implication for astrocytes 4. sRAGE treatment beneficial |
1. Increased RAGE and ligands 2. Decreased sRAGE |
| Multiple Sclerosis | rs2070600 RAGE promoter |
1. Increased RAGE and ligands 2. sRAGE is protective for lifespan and peripheral infiltration 3. Most controversial due to lack of protection in global AGER KO |
1. Lower sRAGE 2. RAGE and ligands increased in disease and MS lesions 3. Ligands go down with disease modifying drugs |
Table 1: The manifestations of AD, PD, ALS and MS are distinct in nature.
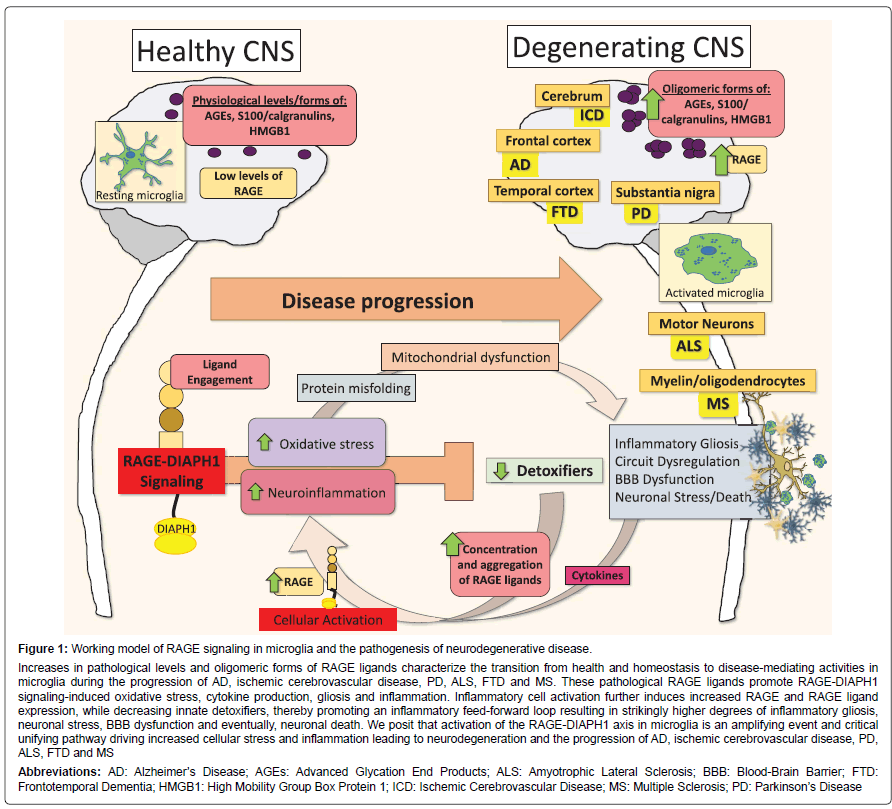
Figure 1: Working model of RAGE signaling in microglia and the pathogenesis of neurodegenerative disease.
Increases in pathological levels and oligomeric forms of RAGE ligands characterize the transition from health and homeostasis to disease-mediating activities in microglia during the progression of AD, ischemic cerebrovascular disease, PD, ALS, FTD and MS. These pathological RAGE ligands promote RAGE-DIAPH1 signaling-induced oxidative stress, cytokine production, gliosis and inflammation. Inflammatory cell activation further induces increased RAGE and RAGE ligand expression, while decreasing innate detoxifiers, thereby promoting an inflammatory feed-forward loop resulting in strikingly higher degrees of inflammatory gliosis, neuronal stress, BBB dysfunction and eventually, neuronal death. We posit that activation of the RAGE-DIAPH1 axis in microglia is an amplifying event and critical unifying pathway driving increased cellular stress and inflammation leading to neurodegeneration and the progression of AD, ischemic cerebrovascular disease, PD, ALS, FTD and MS
Abbreviations: AD: Alzheimer’s Disease; AGEs: Advanced Glycation End Products; ALS: Amyotrophic Lateral Sclerosis; BBB: Blood-Brain Barrier; FTD: Frontotemporal Dementia; HMGB1: High Mobility Group Box Protein 1; ICD: Ischemic Cerebrovascular Disease; MS: Multiple Sclerosis; PD: Parkinson’s Disease
A new age in science is upon us where we are poised to integrate these varied questions. Excitingly, the novel discoveries that have revealed the genetics of disease susceptibility have occurred while many laboratories are concurrently flourishing in their revelations on the cellular and molecular mechanisms of RAGE signal transduction and novel fields have developed to optimize cell targeting and isolation technology, RAGE inhibitors, and more nuanced approaches for clinical trials. Does this mounting evidence suggest a prominent role for RAGE signal transduction in accelerating the pathogenesis of inflammatory neurodegeneration, irrespective of the disease subtype? Further work will undoubtedly be required to determine to what extent and in which specific contexts inhibiting RAGE signaling will protect the CNS from neurodegeneration. However, these developing studies have shown clear benefits of RAGE abrogation, and the future shows promise, particularly as we begin to take a more integrative approach to understanding the complex mechanisms of these devastating diseases and the possibilities of relief through meaningful interventions.
Acknowledgement
We thank Ms. Latoya Woods for the expert assistance in preparation of this review. In addition, we are appreciative of our funding organizations: The National Institute of Health (National Institute on Aging), U.S. Department of Defense, and the Alzheimer’s Association.
References
- Xu Y, Toure F, Qu W, Lin L, Song F, et al. (2010) Advanced glycation end product (AGE)-receptor for AGE (RAGE) signaling and up-regulation of Egr-1 in hypoxic macrophages. J Biol Chem 285: 23233-23240.
- Walker D, Lue LF, Paul G, Patel A, Sabbagh MN (2015) Receptor for advanced glycation endproduct modulators: A new therapeutic target in Alzheimer's disease. Expert Opin Investig Drugs 24:393-399.
- Serrano A, Donno C, Giannetti S, Perić M, Andjus P, et al. (2017) The Astrocytic S100B Protein with Its Receptor RAGE Is Aberrantly Expressed in SOD1(G93A) Models, and its inhibition decreases the expression of proinflammatory genes. Mediators Inflamm 2017: 1626204.
- Schmidt AM, Yan SD, Brett J, Mora R, Nowygrod R, et al. (1993) Regulation of human mononuclear phagocyte migration by cell surface-binding proteins for advanced glycation end products. J Clin Invest 91: 2155-2168.
- Yan SD, Chen X, Schmidt AM, Brett J, Godman G, et al. (1994) Glycated tau protein in Alzheimer disease: A mechanism for induction of oxidant stress. Proc Natl Acad Sci USA 91: 7787-7791.
- Yan SD, Chen X, Fu J, Chen M, Zhu H, et al. (1996) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature 382: 685-691.
- Kislinger T, Fu C, Huber B, Qu W, Taguchi A, et al. (1999) N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem 274: 31740-31749.
- Rai V, Touré F, Chitayat S, Pei R, Song F, et al. (2012) Lysophosphatidic acid targets vascular and oncogenic pathways via RAGE signaling. J Exp Med 209: 2339-2350.
- Giri R, Shen Y, Stins M, Du Yan S, Schmidt AM, et al. (2000) beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am J Physiol Cell Physiol 279: C1772-C1781.
- Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, et al. (2001) Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol 171: 29-45.
- Mi W, Pawlik M, Sastre M, Jung SS, Radvinsky DS, et al. (2007) Cystatin C inhibits amyloid-beta deposition in alzheimer's disease mouse models. Nat Genet 39: 1440-1442.
- Fang F, Lue LF, Yan S, Xu H, Luddy JS, et al. (2010) RAGE-dependent signaling in microglia contributes to neuroinflammation, Abeta accumulation, and impaired learning/memory in a mouse model of alzheimer's disease. FASEB J 24: 1043-1055.
- Morales-Corraliza J, Schmidt SD, Mazzella MJ, Berger JD, Wilson DA, et al. (2013) Immunization targeting a minor plaque constituent clears beta-amyloid and rescues behavioral deficits in an Alzheimer's disease mouse model. Neurobiol Aging 34: 137-145.
- Chauhan G, Adams HHH, Bis JC, Weinstein G, Yu L, et al. (2015) Association of Alzheimer's disease GWAS loci with MRI markers of brain aging. Neurobiol Aging 36: 1765 e7-1765 e16.
- Tominaga T, Sahai E, Chardin P, McCormick F, Courtneidge SA, et al. (2000) Diaphanous-related formins bridge Rho GTPase and Src tyrosine kinase signaling. Mol Cell 5: 13-25.
- Hudson B, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, et al. (2008) Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem 283: 34457-34468.
- Manigrasso MB, Pan J, Rai V, Zhang J, Reverdatto S, et al. (2016) Small molecule inhibition of ligand-stimulated RAGE-DIAPH1 signal transduction. Scientific Reports 6: 22450.
- Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, et al. (2010) Structural basis for ligand recognition and activation of RAGE. Structure 18: 1342-1352.
- Park H, JC Boyington, Adsit FG (2010) The 1.5 Ã… crystal structure of human receptor for advanced glycation endproducts (RAGE) ectodomains reveals unique features determining ligand binding. J Biol Chem 285: 40762-40770.
- Lander HM (1997) Activation of the receptor for advanced glycation end products triggers a p21 ras -dependent mitogen-activated protein kinase pathway regulated by oxidant stress. Journal of Biological Chemistry 272: 17810-17814.
- McDonald DR, Bamberger ME, Combs CK, Landreth GE (1998) β-Amyloid fibrils activate parallel mitogen-activated protein kinase pathways in microglia and THP1 monocytes. J Neurosci 18: 4451-4460.
- He M, Kubo H, Morimoto K, Fujino N, Suzuki T, et al. (2011) Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep 12: 358-364.
- Westerterp M, Murphy AJ, Wang M, Pagler TA, Vengrenyuk Y, et al. (2013) Deficiency of ABCA1 and ABCG1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ Res 112: 1456-1465.
- Daffu G, Shen X, Senatus L, Thiagarajan D, Abedini A, et al. (2015) RAGE suppresses ABCG1-mediated macrophage cholesterol efflux in diabetes. Diabetes 64: 4046-4060.
- Arancio O, Zhang HP, Chen X, Lin C, Trinchese F, et al. (2004) RAGE potentiates Abeta-induced perturbation of neuronal function in transgenic mice. EMBO J 23: 4096-4105.
- Lo MC, Chen MH, Lee WS, Lu CI, Chang CR, et al. (2015) Nε-(carboxymethyl) lysine-induced mitochondrial fission and mitophagy cause decreased insulin secretion from β-cells. Am J Physiol Endocrinol Metab 309: E829-E839.
- Kang R, Tang D, Schapiro NE, Loux T, Livesey KM, et al. (2014) The HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by regulating mitochondrial bioenergetics. Oncogene 33: 567-577
- Wang X, Yu S, Wang CY, Wang Y, Liu HX, et al. (2015) Advanced glycation end products induce oxidative stress and mitochondrial dysfunction in SH-SY5Y cells. In Vitro Cell Dev Biol Anim 51: 204-209.
- Tsoporis JN, Izhar S, Leong-Poi H, Desjardins JF, Huttunen HJ, et al. (2010) S100B Interaction with the receptor for advanced glycation end products (RAGE). Circ Res 106: 93-101.
- Yoshimaru T, Suzuki Y, Inoue T, Nishida S, Ra C, et al. (2008) Extracellular superoxide released from mitochondria mediates mast cell death by advanced glycation end products. Biochim Biophys Acta 1783: 2332-2343.
- Iwamoto K, Kanno K, Hyogo H, Yamagishi S, Takeuchi M, et al. (2008) Advanced glycation end products enhance the proliferation and activation of hepatic stellate cells. J Gastroenterol 43: 298-304.
- Coughlan MT, Thorburn DR, Penfold SA, Laskowski A, Harcourt BE, et al. (2009) RAGE-induced cytosolic ROS promote mitochondrial superoxide generation in diabetes. J Am Soc Nephrol 20: 742-752.
- Basta G, Lazzerini G, Turco SD, Ratto GM, Schmidt AM, et al. (2005) At least 2 distinct pathways generating reactive oxygen species mediate vascular cell adhesion molecule-1 induction by advanced glycation end products. arteriosclerosis, thrombosis, and vascular biology. Arterioscler Thromb Vasc Biol 25: 1401-1407.
- Posey SC, BE Bierer (1999) Actin stabilization by jasplakinolide enhances apoptosis induced by cytokine deprivation. J Biol Chem 274: 4259-4265.
- Fukata M, Nakagawa M, K Kaibuchi (2003) Roles of Rho-family GTPases in cell polarisation and directional migration. Curr Opin Cell Biol 15: 590-597.
- Toure F, Fritz G, Li Q, Rai V, Daffu G, et al. (2012) Formin mDia1 mediates vascular remodeling via integration of oxidative and signal transduction pathways. Circ Res 110: 1279-1293.
- Zhang C, Wang L, Chen J, Liang J, Xu Y, et al. (2017) Knockdown of Diaph1 expression inhibits migration and decreases the expression of MMP2 and MMP9 in human glioma cells. Biomed Pharmacother 96: 596-602
- Saleh A, Smith DR, Tessler L, Mateo AR, Martens C, et al. (2013) Receptor for advanced glycation end-products (RAGE) activates divergent signaling pathways to augment neurite outgrowth of adult sensory neurons. Exp Neurol 249: 149-159.
- Bucciarelli LG, Ananthakrishnan R, Hwang YC, Kaneko M, Song F, et al. (2008) RAGE and modulation of ischemic injury in the diabetic myocardium. Diabetes 57: 1941-1951.
- Rouhiainen A, Kuja Panula J, Wilkman E, Pakkanen J, Stenfors J, et al. (2004) Regulation of monocyte migration by amphoterin (HMGB1). Blood 104: 1174-1182
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, et al. (2013) TREM2 variants in alzheimer's disease. N Engl J Med 368: 117-127.
- Jonsson T, Stefansson H, Stacy Steinberg, Jonsdottir I, Palmi V. Jonsson, et al. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368: 107-116.
- Hickman SE, JE Khoury (2014) TREM2 and the neuroimmunology of alzheimer's disease. Biochem Pharmacol 88: 495-498.
- Abuznait AH, A Kaddoumi (2012) Role of ABC transporters in the pathogenesis of Alzheimer's disease. ACS Chem Neurosci 3: 820-831.
- Villegas Llerena C, Phillips A, Garcia Reitboeck P, Hardy J, Pocock JM (2015) Microglial genes regulating neuroinflammation in the progression of Alzheimer's disease. Curr Opin Neurobiol 36: 74-81.
- Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, et al. (2012) ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science 335: 1503-1506.
- Zhang ZG, Li Y, Ng CT, Song YQ (2015) Inflammation in alzheimer's disease and molecular genetics: recent update. Arch Immunol Ther Exp (Warsz) 63: 333-344.
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, et al. (2013) Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496: 238-242.
- Barzilay JI, Jablonski KA, Fonseca V, Shoelson SE, Goldfine AB, et al. (2014) The impact of salsalate treatment on serum levels of advanced glycation end products in type 2 diabetes. Diabetes Care 37: 1083-1091.
- Ransohoff RM, Perry VH (2009) Microglial physiology: Unique stimuli, specialized responses. Annu Rev Immunol 27: 119-145.
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, et al. (2010) CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol 177: 2549-2562.
- Saederup N, Cardona AE, Croft K, Mizutani M, Cotleur AC, et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 5: e13693.
- Prinz M, Priller J, Sisodia SS, Ransohoff RM (2011) Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat Neurosci 14: 1227-1235.
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, et al. (2014) Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17: 131-143.
- Katsumoto A, Lu H, Miranda AS, Ransohoff RM (2014) Ontogeny and functions of central nervous system macrophages. J Immunol 193: 2615-2621.
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, et al. (2015) Neuroinflammation in alzheimer's disease. Lancet Neurol 14: 388-405.
- Ransohoff RM, J El Khoury (2015) Microglia in Health and Disease. Cold Spring Harb Perspect Biol 8: a020560.
- Khoury J, Hickman SE, Thomas CA, Loike JD, Silverstein SC, et al. (1998) Microglia, scavenger receptors, and the pathogenesis of Alzheimer's disease. Neurobiol Aging 19: S81-S84.
- Hickman SE, Kingery ND, Ohsumi T, Borowsky M, Wang LC, et al. (2013) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16: 1896-1905.
- Schafer Dorothy P, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, et al. (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74: 691-705.
- Bilimoria PM, Stevens B(2015) Microglia function during brain development: New insights from animal models. Brain Res 1617: 7-17.
- Schafer DP, Stevens B (2015) Microglia function in central nervous system development and plasticity. Cold Spring Harb Perspect Biol 7: a020545
- Wu Y, Dissing-Olesen L, MacVicar BA, Stevens B, et al. (2015) Microglia: Dynamic mediators of synapse development and plasticity. Trends Immunol 36: 605-613.
- Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, et al. (2009) RAGE-mediated signaling contributes to intraneuronal transport of amyloid-β and neuronal dysfunction. Proc Natl Acad Sci U S A 106: 20021-20026.
- Kuhla B, Boeck K, Schmidt A, Ogunlade V, Arendt T, et al. (2007) Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer's disease brains. Neurobiol Aging 28: 29-41.
- More SS, Vartak AP, Vince R (2013) Restoration of glyoxalase enzyme activity precludes cognitive dysfunction in a mouse model of alzheimer’s disease. ACS Chem. Neurosci 4: 330-338.
- Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, et al. (2004) Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 113: 1641-1650.
- Gasparotto J, Girardi CS, Somensi N, Ribeiro CT, Moreira JCF, et al. (2017) Receptor for advanced glycation endproducts mediates sepsis-triggered amyloid-β accumulation, tau phosphorylation, and cognitive impairment. J Biol Chem 293:226-244
- Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, et al. (2009) Systemic inflammation and disease progression in Alzheimer disease. Neurology 73: 768-774.
- Wang ZX, Wan Y, Tan L, Liu J, Wang HF, et al. (2017) Genetic association of HLA gene variants with MRI brain structure in alzheimer’s disease. Mol Neurobiol 54: 3195-3204.
- Hofmann MA, Drury S, Hudson BI, Gleason MR, Qu W, et al. (2002) RAGE and arthritis: The G82S polymorphism amplifies the inflammatory response. Genes Immun 3: 123-135.
- Li K, Zhao B, Dai D, Yao S, Liang W, et al. (2011) A functional p.82G>S polymorphism in the RAGE gene is associated with multiple sclerosis in the Chinese population. Mult Scler 17: 914-921.
- Miller S, Henry AP, Hodge E, Kheirallah AK, Billington CK, et al. (2016) The ser82 RAGE variant affects lung function and serum RAGE in smokers and sRAGE production in vitro. PLoS One 11: e0164041.
- Wang Y, Wu L, Li J, Fang D, Zhong C, et al. (2015) Synergistic exacerbation of mitochondrial and synaptic dysfunction and resultant learning and memory deficit in a mouse model of diabetic Alzheimer's disease. J Alzheimers Dis 43: 451-463.
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, et al. (2003) RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9: 907-913.
- Qosa H, LeVine H , Keller JN, Kaddoumi A (2014) Mixed oligomers and monomeric amyloid-β disrupts endothelial cells integrity and reduces monomeric amyloid-β transport across hCMEC/D3 cell line as an in vitro blood–brain barrier model. Biochim Biophys Acta 1842: 1806-1815.
- Burstein A, Grimes I, Galasko DR, Aisen PS, Sabbagh M, et al. (2014) Effect of TTP488 in patients with mild to moderate alzheimer's disease. BMC Neurol 14: 12.
- Galasko D, Bell J, Mancuso JY, Kupiec JW, Sabbagh MN, et al. (2014) Clinical trial of an inhibitor of RAGE-Abeta interactions in Alzheimer disease. Neurology 82: 1536-1542.
- Ma WQ, Qu QR, Zhao Y, Liu NF (2016) Association of RAGE gene Gly82Ser polymorphism with coronary artery disease and ischemic stroke: A systematic review and meta-analysis. Medicine 95: e5593.
- Tang SC, Yeh SJ, Tsai LK, Hu CJ, Lien LM, et al. (2016) Cleaved but not endogenous secretory RAGE is associated with outcome in acute ischemic stroke. Neurology 86: 270-276.
- Greco R, Demartini C, Zanaboni AM, Blandini F, Amantea D,et al. (2017) Modulation of cerebral RAGE expression following nitric oxide synthase inhibition in rats subjected to focal cerebral ischemia. Eur J Pharmacol 800: 16-22.
- Harada S, Matsuura W, Liu K, Nishibori M, Tokuyama S (2016) Possible involvement of the HMGB1/RAGE signaling mechanism in the induction of central post-stroke pain induced by acute global cerebral ischemia. Brain Res 1646: 433-440.
- Liesz A, Dalpke A, Mracsko E, Antoine DJ, Roth S, et al. (2015) DAMP Signaling is a key pathway inducing immune modulation after brain injury. J Neurosci 35: 583-598.
- Khan MA, Schultz S, Othman A, Fleming T, Lebrón-Galán R, et al. (2016) Hyperglycemia in stroke impairs polarization of monocytes/macrophages to a protective noninflammatory cell type. J Neurosci 36: 9313-9325.
- Hu J, Liu B, Zhao Q, Jin P, Hua F, et al. (2016) Bone marrow stromal cells inhibits HMGB1-mediated inflammation after stroke in type 2 diabetic rats. Neuroscience 324: 11-19.
- Origlia N, Criscuolo C, Arancio O, Yan SS, Domenici L (2014) RAGE inhibition in microglia prevents ischemia-dependent synaptic dysfunction in an amyloid-enriched Environment. J Neurosci 34: 8749-8760.
- Goetz CG (2011) The history of parkinson's disease: Early clinical descriptions and neurological therapies. Cold Spring Harb Perspect Med 1: a008862.
- Oliveira SA, Scott WK, Nance MA, Watts RL, Hubble JP, et al. (2003) Association study of parkin gene polymorphisms with idiopathic parkinson disease. Arch Neurol 60: 975-980.
- Gasparotto J, Ribeiro CT, Bortolin RC, Somensi N, Rabelo TK, et al. (2017) Targeted inhibition of RAGE in substantia nigra of rats blocks 6-OHDA–induced dopaminergic denervation. Sci Rep 7: 8795.
- Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, et al. (2012) S100B is increased in parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-α pathway. Brain 135: 3336-3347.
- Teismann P, Sathe K, Bierhaus A, Leng L, Martin HL, et al. (2012) Receptor for advanced glycation endproducts (RAGE) deficiency protects against MPTP toxicity. Neurobiol Aging 33: 2478-2490.
- Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, et al. (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3: 17085.
- Kikuchi S, Shinpo K, Ogata A, Tsuji S, Takeuchi M, et al. (2002) Detection of N epsilon-(carboxymethyl)lysine (CML) and non-CML advanced glycation end-products in the anterior horn of amyotrophic lateral sclerosis spinal cord. Amyotroph Lateral Scler Other Motor Neuron Disord 3: 63-68.
- Juranek JK, Daffu GK, Wojtkiewicz J, Lacomis D, Kofler J, et al. (2015) Receptor for advanced glycation end products and its inflammatory ligands are upregulated in amyotrophic lateral sclerosis. Front Cell Neurosci 9: 485.
- Casula M, Iyer AM, Spliet WG, Anink JJ, Steentjes K, et al. (2011) Toll-like receptor signaling in amyotrophic lateral sclerosis spinal cord tissue. Neuroscience179: 233-243.
- Kaufmann E, Boehm BO, Süssmuth SD, Kientsch-Engel R, Sperfeld A, et al. (2004) The advanced glycation end-product N epsilon-(carboxymethyl)lysine level is elevated in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Neurosci Lett 371: 226-229.
- Ilzecka J (2009) Serum-soluble receptor for advanced glycation end product levels in patients with amyotrophic lateral sclerosis. Acta Neurol Scand 120: 119-122.
- Juranek JK, Daffu GK, Geddis MS, Li H, Rosario R, et al. (2016) Soluble RAGE treatment delays progression of amyotrophic lateral sclerosis in SOD1 mice. Front Cell Neurosci 10: 117.
- Lo Coco D, Veglianese P, Allievi E, Bendotti C (2007) Distribution and cellular localization of high mobility group box protein 1 (HMGB1) in the spinal cord of a transgenic mouse model of ALS. Neurosci Lett 412: 73-77.
- Kim MJ, Vargas MR, Harlan BA, Killoy KM, Ball LE,et al. (2017) Nitration and glycation turn mature NGF into a toxic factor for motor neurons: A role for p75(NTR) and RAGE signaling in ALS. Antioxid Redox Signal
- Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, et al. (2017) Amyotrophic lateral sclerosis. Nat Rev Dis Primers 3:17085.
- O’Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, et al. (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science 351: 1324-1329.
- Nilson AN, Kelsey C, Gerson JE, Whittle TB, Crain CN, et al. (2016) Tau oligomers associate with inflammation in the brain and retina of tauopathy mice and in neurodegenerative diseases. J Alzheimers Dis 55: 1083-1099.
- Green AJE, Harvey RJ, Thompson EJ, Rossor MN (1997) Increased S100β in the cerebrospinal fluid of patients with frontotemporal dementia. Neurosci Lett 235: 5-8.
- Caillier SJ, Briggs F, Cree BA, Baranzini SE, Fernandez-Viña M, et al. (2008) Uncoupling the roles of HLA-DRB1 and HLA-DRB5 genes in multiple sclerosis. J Immunol 181: 5473-5480.
- Tiszlavicz Z, Gyulai Z, Bencsik K, Szolnoki Z, Kocsis AK, et al. (2009) RAGE gene polymorphisms in patients with multiple sclerosis. J Mol Neurosci 39: 360-365.
- Sternberg Z, Weinstock Guttman B, Hojnacki D, Zamboni P, Zivadinov R, et al. (2008) Soluble receptor for advanced glycation end products in multiple sclerosis: a potential marker of disease severity. Mult Scler 14: 759-763.
- Andersson A, Covacu R, Sunnemark D, Danilov AI, Dal Bianco A, et al. (2008) Pivotal advance: HMGB1 expression in active lesions of human and experimental multiple sclerosis. J Leukoc Biol 84: 1248-1255.
- Barateiro A, Afonso V, Santos G, Cerqueira JJ, Brites D, et al. (2016) S100B as a potential biomarker and therapeutic target in multiple sclerosis. Mol Neurobiol 53: 3976-3991.
- Sternberg Z, Sternberg D, Chichelli T, Drake A, Patel N,et al. (2016) High-mobility group box 1 in multiple sclerosis. Immunol Res 64: 385-391.
- Sternberg Z, Kolb C, Chadha K, Nir A, Nir R, et al. (2017) Fingolimod anti-inflammatory and neuroprotective effects modulation of RAGE axis in multiple sclerosis patients. Neuropharmacology 130: 71-76.
- Yan SS, Wu ZY, Zhang HP, Furtado G, Chen X, et al. (2003) Suppression of experimental autoimmune encephalomyelitis by selective blockade of encephalitogenic T-cell infiltration of the central nervous system. Nat Med 9: 287-293.
Citation: Derk J, MacLean M, Juranek J, Schmidt AM (2018) The Receptor for Advanced Glycation Endproducts (RAGE) and Mediation of Inflammatory Neurodegeneration. J Alzheimers Dis Parkinsonism 8: 421. DOI: 10.4172/2161-0460.1000421
Copyright: ©2018 Derk J, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 9219
- [From(publication date): 0-2018 - Dec 20, 2025]
- Breakdown by view type
- HTML page views: 8141
- PDF downloads: 1078