Research Article Open Access
Unusual Post-Spray Proton Transfer to Protein Using Acetone Spray in Desorption Electrospray Ionization
Anna Warnet1*, Nicolas Auzeil2 and Jean-Claude Tabet1
1CSOB-Institut Parisien de Chimie Moléculaire (UMR 8232-UFR 926), CNRS, Université Pierre et Marie Curie, Paris, France
2EA4463: Laboratoire de chimie et toxicologie analytique et cellulaire, Université Paris Descartes, Faculté des Sciences Pharmaceutiques 4 avenue de l’Observatoire, Paris, France
- *Corresponding Author:
- Anna Warnet
CSOB-Institut Parisien de Chimie Moléculaire (UMR 8232-UFR 926)
CNRS, Université Pierre et Marie Curie (UPMC), 75005 Paris, France
Tel: 33-0-1-44-27-518
E-mail: anna.warnet@upmc.fr
Received date: August 26, 2015; Accepted date: October 01, 2015; Published date: October 07, 2015
Citation: Warnet A, Auzeil N, Tabet JC (2015) Unusual Post-Spray Proton Transfer to Protein Using Acetone Spray in Desorption Electrospray Ionization. J Anal Bioanal Tech 6:283. doi:10.4172/2155-9872.1000283
Copyright: © 2015 Warnet A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Analytical & Bioanalytical Techniques
Abstract
Although acetone, in DESI ionisation generally leads to protein aggregation, in this study we report unexpected multi-proton transfers to lysozyme using this aprotic solvent as a charged spray. The DESI/acetone mass spectrum of lysozyme displays (i) a significant increase in the average charge state (Zav) and (ii) an incomplete H+/Ca2+ exchange, even though the overall contribution of cationised species is high, relative to those from spraying with a methanol/water solvent. This behavior is contrary to that expected from gas phase basicity, because GBacetone >GBmethanol. Decreasing the amount of sample deposited on the target (from 50 to 0.050 pmole) leads to a charge state increase, as seen in ESI, but not in the extent of cationisation. Moreover, the DESI signal duration is extended with sprayed acetone even though the total ionic current is significantly lowered. With a d6-acetone spray, no incorporation of a deuteron occurs, and the ionization yield is strongly decreased for multi-protonated lysoi+ lysozyme. This is in contrast to that observed with a d4-methanol spray, which displays a distribution of 48 deuterons in the lyso9+ ion as shown in high resolution with a LTQ/Orbitrap instrument. This unexpected behavior of the (CD3)2CO spray suggests that protons do not originate from acetone. Furthermore, dry argon post-flow on the target surface results in the lysozyme signal suppression, whereas with a humid argon flow, the signal is regenerated. On the other hand, an argon stream bubbling in heavy water, yields incorporation of several deuterons. The interpretation of this behavior is explained by considering the acetone radical ions at the surface of the primary droplets (and/or offspring droplets and/or at the wet sample surface), being able to react with ambient moisture (or with traces of water adsorbed at liquid phase). Under these conditions, enough protons are produced to generate multi-charged solvated lysozyme aggregates which then become desolvated in the reduced pressure in the skimmer area.
Keywords
MS; DESI; Aprotic solvent; Lysozyme; Cationization; Labelling
Introduction
Desorption electrospray ionization mass spectrometry (DESIMS), an ambient ionization method first reported by Takats et al. in 2004 [1], permits fast analysis of bulk samples without pre-treatment. Many applications appeared rapidly [1-6] and their number still increases [7]. Since, over forty ambient ionization methods have now been developed, based on sprays with [1] or without high voltage [8], acoustic mode [9-12], chemical ionization [10], laser [13], plasmas [14], heating [15] and combined techniques [6]. Several of these ambient ionization modes can lead to imaging analysis [16]. Based on the extractive approaches [7], they permit monitoring reaction [17] and intermediate studies in fast reactive processes [18-20]. Furthermore, it is possible to combine them with an electrochemistry device [21,22]. Among the various ambient ionization/desorption modes, DESI-MS [1] evolved as the most popular mode and is suitable for analysis of a large panel of organic compounds. It has been reported in a wide area of applied analyses including forensic [3-6,8,23,24], homeland security [4,5,25-28], food contaminants [29] and agrochemicals [30,31], metabolites [4,32-34], drugs, and drugs of abuse [34-39]. DESI mode also contributes to biomolecule detection [40-46], as well as direct analysis of heterogeneous biological material [3,4,47-50] and in imaging [40,48,51]. For larger analytes, DESI produces efficiently multicharged proteins [44,52-56] as well as non-covalent complexes [54,57-62].
The proposed DESI mechanism based on the sample extraction from a thin target layer and transmission solvated ionic species to the transfer capillary, known as “droplet pick-up” [5,7,63], may be regarded as a process of four distinct steps occurring from the microelectrospray source assisted by auxiliary gas flow and yielding charged solvent micro-sized droplets (d ≤ 10 μm). As described in literature [1-5], these steps involve: (i) the impacting of the charged droplets to the target surface with velocities of the order of 100 m/s at an angle ranging from 20 to 90 degrees, (ii) the wetting and subsequent partial dissolution of the sample deposited on the target, (iii) the momentum transfer from the multiple impacting droplets, originating from a shock wave-like phenomenon [4], which leads to production of analyte offspring droplets (characterized by a distribution in sizes and in velocities) from target surface layer to move toward the transfer capillary. Those droplets are significantly smaller than those generated in ESI before the droplet Coulomb explosions [64] (except for a large for which mainly the “charge residue model” mechanism must be considered [65]), (iv) Finally, this is followed by the release of solvated analyte ions, similar in charge but probably smaller than those generated in the ESI process [32]. The DESI mechanism briefly presented above specially emphasizes the importance of: (i) properties of the surface used for the sample deposition, and (ii) properties of the selected electrospray solvent and additives to enhance ionization efficiency [66,67]. The surface must have a weak affinity for analyte [68] to facilitate its extraction/dissolution in the formed thin solvent layer. In contrast, generally a high affinity between the surface and the sprayed solvent increases the signal intensity and its stability. Moreover, due to the ESI electrical field, the insulated surface undergoes electrostatic charge closely related to its conductivity, and is responsible for the microdroplet ejection from the thin solvent layer [68-70].
Depending on the analyte, one can use various surfaces for sample deposition. The most popular materials in analysis of protein, carbohydrates, and synthetic organic compounds are: polymethylmethacrylate (PMMA) [42,43,71], polytetra-fluoro-ethylene (PTFE) [1,42,71-73], glass [42,71,74], and paper [71,74] (different to the Paper Spray mode, another ambient ionization mode) [75], with a limit of detection (LOD) [26,42-44,68] from 0.1 to 2000 pg.mm-2. More recently, nanoporous silicon and ultra-thin layer chromatography UTLC led to improve LOD values compared with PMMA and PTFE surfaces [68]. Proteomic analysis uses commonly nanoporous alumina surface [76].
The sprayed solvent is also a crucial parameter in the DESI process [7,59]. In fact, polarity, boiling point and viscosity are the major macroscopic properties that affect DESI performances [66,67]. When it is required, other properties as volatility and capacity to dissolve the sample, guide towards particular solvents. Stability and thickness of the solvent layer, correlated with intensity and stability of the signal depend on the analyte interactions with the surface and the evaporation rate. Moreover, the solvent polarity influences dissolution efficiency in solvent layer, and hence analyte concentration in the offspring droplet evaporation [26,69,77]. It is known that protic solvents favor the stabilization of the analyte charge during the offspring droplet evaporation, although using non-aqueous solvents is possible if, at first, the analyte is dissolved. Indeed that method generates ions containing weaker internal energies [66,67]. In practice, the most commonly employed solvent in DESI-MS consists of a methanol-water mixture [1]. However, depending on the analyte, an aprotic solvent such as acetonitrile may advantageously replace methanol [30,52,66,67,77].
Furthermore, addition of a small amount (0.1% v/v) of a protonating agent, e.g., formic, acetic, or trifluoroacetic acids, facilitates analyte protonation [72,76]. In some cases, pure solvents were proposed, for instance in the targeting of plant alkaloids separated on a thin-layer chromatography plate. Van Berkel et al. reported higher ionization efficiency with acetonitrile than with methanol [78,79]. Recently, Badu- Tawiah et al. demonstrated that the acetonitrile/chloroform (1/1) or tetrahydrofuran/chloroform (1/1) mixtures exhibit an improved efficiency compared to methanol/ water (1/1) for hydrophobic analytes detected under DESI-MS conditions [66,67]. Note that the addition of particular reagents (e.g., m-nitrobenzyl alcohol (m-NBA) or sulfolane) permits the supercharging of protein analytes [79].
In the present work, we chose oxidized lysozyme as model in order to explore the potentiality of unusual aprotic solvent for desorption of small proteins within an enough efficiency to detect their characteristic ionized species. Its medium size and its multiple disulfide bridges prevent protein denaturation but the large amount of conformational freedom for the protein motivates this choice. Moreover, lysozyme includes 19 basic residues, which promote protonation in DESI. In this work, among the less used aprotic solvents, we selected anhydrous acetone, although it is considered as an inefficient solvent for protein solubilization. We opted for a PTFE target surface because of its hydrophobic character, which manifests strong interactions with acetone and results in stabilization of the thin solvent layer. Therefore, the recorded mass spectra of lysozyme were carefully examined, especially in terms of charge state distribution (CSD) [80] and average charge states Zav [81-83] of cationized and multi-protonated lysozyme species. Systematically, we compared them to those acquired with commonly sprayed protic solvents, e.g., methanol/water. In addition, in order to contribute to understand the details of the ionization mechanism i.e., the origin of ionizing protons when an aprotic solvent such as acetone is used, we modified the DESI ambient conditions by using post-flow gas. We performed DESI experiments by using sprayed anhydrous acetone (labelled or not) either in conventional ambient conditions or combined to an additional post-flow gas: (i) dry argon, and (ii) humidified argon/water (labeled or not).
Experimental
Chemicals and reagents
HPLC grade methanol, d4-methanol (CD3OD) and d6-acetone [(CD3)2CO] were acquired from Sigma (St Quentin Fallavier, France). The d6-acetone was used extemporaneously. HPLC grade acetone and formic acid, were supplied from WWR International (Fontenay-sous- Bois, France), and argon, nitrogen were purchased from Air-Liquide (Nanterre, France). Anhydrous acetone was prepared in accordance to the protocol of Yves Baratoux prior DESI/MS analysis [84]. Water was purified to 18.2 MΩ.cm with a milli-Q water system, from Millipore (El Paso, TX, USA). Hen Egg White lysozyme, supplied by Sigma (Saint- Quentin Fallavier, France), was purified according to the procedure of Thomas et al. [84] and was partially desalted to its isoionic state [85]. Finally, the pH of the isoionic protein solution was adjusted to pH 4.5 with acetic acid, and Lysozyme concentration (approximately 100 μM) was determined using UV-spectrophotometry. This solution was then diluted to appropriate concentrations (0.001 to 100 μM) with pure water, before DESI-MS analysis. Polytetra-fluoro-ethylene surfaces (PTFE plates 1/16 inch, 2.95 inch × 0.98 inch) were purchased from Isoflon SAS (Diemoz, France).
DESI mass spectrometry
DESI-MS experiments were performed using a LTQ OrbitrapTM from Thermo Fisher Scientific (Courtaboeuf, France). The analyser was operated in the FTMS mode (high resolving power fixed at 105). Two micro scans were used to record one scan and the maximum injection time was 0.200 s. DESI mass spectrum acquisition was done from m/z 200 to m/z 4000 (i.e., high m/z ratio range selection mode). XcaliburTM software (Thermo Fisher Scientific) was used for data acquisition and analysis. DESI experiments, in positive ion polarity mode, were carried out using an Omni SprayTM Ion source from Prosolia, Inc. (Indianapolis, IN, USA) equipped with a manual X-Y-Z positioner. A double chargecoupled device (CCD) camera was used for positioning and retaining in place as accurately as possible the deposited sample. The following optimized values for experimental parameters were: spray voltage, 3.8 kV; capillary temperature, 300°C; capillary voltage, 49 V; and tube lens, 250 V. For DESI source, preferred parameters hereafter were: solvent flow rate, 5 μL.min-1; spray angle, 37°; distance from sprayer to PTFE surface, approximately 0.5 mm; distance from sample deposit to mass spectrometer inlet, 1-2 mm; dry nitrogen gas pressure, 72 psi.
In a typical DESI-MS experiment, seven aliquots of aqueous Lysozyme solution (0.5 μL-10 μM) deposited on PTFE plate, were left for approximately 10 min, at room temperature until total dry. Successively, each deposit (approximately 5 pmoles) was analyzed by a manual sweep. Before use, the PTFE plate was sonicated for 5 min in 0.1% aqueous formic acid, rinsed with water and dried under a nitrogen flow. DESI mass spectra are the scan average obtained from the spots.
Notation and thermochemistry
The large ionic species (produced at the end of droplet lifetime), with a large and excess of charge number on the studied protein, stabilized by a lot of solvent molecules, are herein called charged aggregates. However, it does not arise with protein aggregates which are very minor species. The charged aggregates are macromolecular non covalent systems which are desolvated under reduced pressure in the skimmer area [86].
To simplify the ion notation, the multi-protonated lysozyme species with n protons, [Lyso + nH]n+ (or LysoHn n+), were denoted as Lyson+ [87]. The multi-protonated/cationized forms, e.g., [Lyso + (nkN) H + kC]n+ (with C=cation, N=cation valence number, k=number of cations), were denoted as LysoCk n+. The main adduct ion corresponding to an m/z shift relative to that of Lyson+ by 38 k/n m/z (with k=1) is observed. This means that cationization occurs mainly with one (or more) K+ and/or Ca2+ cations. It results the formation either [Lyso + (n-p)H + pK]n+ and/or [Lyso + (n-2q)H + qCa]n+ (p and q as number of alkali and alkaline earth cations, respectively) noted as LysoKp n+ and LysoCaq n+. The average charge state [81-83] corresponding to the following ratio was noted as Zav, with Ii+, related to the charge i, as the sum of the peak intensities I(h)i+ and ΣI(h/c)i+> (h and c, in subscript letters, characterize peak intensities corresponding to ions constituted by protons and metallic cation(s)) of the Lysoi+ and ΣLysoCk i+ ions, respectively :
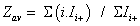
These values are related in particular to the protein conformations (seen supplementary S2 material with included references [88-93] for Lysozyme). Thus, the Zav expression is composed by the sum of two terms (Z(h)av + Z(h/c)av), which were explored mainly when sprayed acetone was used:
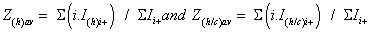
To compare on one hand, the stability of the multi-charged species related to its environment, and on the other hand, the possible proton exchanges with solvent, the apparent gas phase [94,95], GBapp(Lyson+), was used (Equation 1):
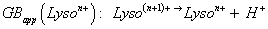 (1)
(1)
This definition is based on the gas phase basicity GB(M) definition, a thermochemical state ΔG° basicity(M) function, the Gibbs energy change related to the [MH+ → M + H+] fictive proton desolvation reaction. Its ΔH° term is called proton affinity PA(M) [95,96] and for multiplied charged Lysozyne, this term is PAapp(Lyso(n-1)+) [91,92] (with some of their GBapp values from literature are provided) [83,97-102].
By analogy to PAapp(Lyson+), apparent alkaline earth cation affinity (e.g., for Ca2+, cation chosen for our data, seen supplementary material S3 with included references [104-112]) of multi-protonated Lyso(n-1)+ species is noted as CaCAapp(Lyso(n-1)+) and CaCBapp(Lyso(n-1)+) for the Gibbs energy change of Equation 2 [103], whereas the gas phase basicity of cationized multi-protonated LysoCan+ Lysozyme noted as GBapp(LysoCan+) (Equation 3), are considered to be:
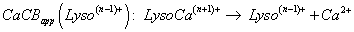 (2)
(2)
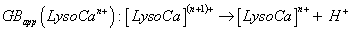 (3)
(3)
The GBapp state function is introduced from the CB cation affinity particularly known for amino-acid neutrals (AA) [104-108].
Results and Discussion
As previously stated, the solvent is one of important factors [7,66,67] among several experimental parameters concerned about CSD (seen experimental part and supplementary material S2) of ions in gas phase, with a maximum charge state of solvent-free proteins, resulting from desolvation of multi-charged aggregates in ESI mode. Indeed, it implies at the same time, the droplet charge evolution by macroscopic effects, and intra-aggregate protons/alkali (or alkaline earth) cations exchange reactions by macromolecular effects. The macroscopic effects influence the surface tension, and thus, the droplet size/shape, their analyte concentration, and the formation of large charged aggregates either by “ion evaporation” [113-115] desorbed or produced from “charged residue” process [65]. The macromolecular effects and gas phase thermochemistry, act on the CSD, as on the maximum of the charge state of the multiply-charged solvent-free proteins. The situation somewhat differs in DESI mode, because the charged offspring droplets are significantly smaller and distorted in a shell-shape due to their high velocity [2,4,63,65,67]. This results in higher speed favoring their fast fission through the produced dragging force.
Despite these minor differences, Myung et al. [43] demonstrated similar charge distributions in both the ESI and DESI modes in ion mobility experiments. This may involve a compensation of the macroscopic and macromolecular effects, which relative importance varies with the mode of desorption, resulting in similar ESI (2.1 μM concentration of the used lysozyme solution) and DESI mass spectra (recorded from 5 pmoles of deposed lysozyme). Although, the discussion of the macroscopic effect influence on the dynamic of plume should deserve a fundamental interest, this aspect will not be discussed in this study any more. Actually, the processes involved in the aggregate ion formation from the offspring droplets have not been particularly scrutinized in contrast to the role of the agent providing available protons for charging the droplets (as with the charged aggregates).
In order to investigate the solvent role in providing charges, we studied the influence of the lysozyme amount in the offspring droplets on CSD and Zav of Lyson+ (and LysoCk n+), using first sprayed protic solvent (methanol/water mixture). Besides, distribution of cations (e.g., Na+, and/or K+ …) was notably scrutinized, according to lysozyme CSD, just like the deposited lysozyme amount. Secondarily, the effects, while changing sprayed protic solvent by an aprotic as acetone (an inappropriate solvent for lysozyme) were explored on the previous characteristics (i.e., CSD, Zav and cation distribution).
CSD and Zav values for Lyson+ produced in DESI-MS from sprayed aqueous protic solvent
Under DESI conditions with acidified methanol/water mixture (8/2) (Figure 1a) with 5 pmole of deposited lysozyme, three peaks of the lysozyme mass spectrum dominate at m/z 1431.49, m/z 1590.45, and m/z 1789.12, corresponding to ions with 10+, 9+ and 8+ charge states, respectively. Consequently, a narrow CSD value (i.e., from 7+ to 11+) characterizes this profile, with a calculated average charge state as Zav=(9.0 ± 0.1), which is slightly lower than that obtained in ESI i.e., Zav=(9.8 ± 0.1) for the lysozyme sample consumption considered approximately similar (Supplementary material S1 and Figure S1) in both the experiments (i.e., instrument, in skimmer desolvation and ion transmission conditions). Takas et al. reported already this behavior [4]. The weak differences observed between these ESI and DESI experiments may signify that the yield of ionization/desorption are not very different, although the latter is somewhat gentler than the former. This result is consistent with those provided from the study of Myung et al. [43], which shown similar conclusion from IMS experiments performed using sprayed protic solvents.

Figure 1: DESI mass spectra of 5 pmoles of deposited lysozyme on PTFE target with spray beam prepared (a) with methanol/water (8/2 v/v) mixture and 0.1% formic acid, and (b) pure anhydrous acetone. In inset of each mass spectrum, a zoom of the 8+ charge state species (i.e., Lyso8+ and its cationized forms noted as LysoCk8+).
For the production of solvent-free multi-charged lysozyme Lyson+ species, it can be assumed that formation of the multi-protonated aggregates occurs from the small offspring droplets followed by “in skimmer” desolvation steps, as it is described by an ESI-like mechanism [4]. However, to explain the origin of the small variation of Zav between the profiles of ESI (Supplementary material S1 and Figure S1) and DESI (Figure 1), the net charge carried by lysozyme/solvent aggregates has been qualitatively scrutinized by considering the Lyson+ ions.
In DESI mode, the Zav decrease was previously ascribed to the small sizes of the offspring droplets [64,68]. Very likely, these latter carry a charge number lower than those produced in ESI. This led to a positive net charge decrease on the solvated and folded protein surface, and thus, after its solvent release from the charged lysozyme/ solvent aggregates, the solvent-free ionized proteins are characterized by a slightly reduced average charge state. On the other hand, this slight Zav discrepancy between the ESI and DESI mass spectra may be due to the nature (protons vs. cations) of the charge transfer mechanism of DESI overall process, in which protic solvent properties do not strongly affect both the ionization steps and production of solvent-free protein ions [43,67,80]. This interpretation may rationalize the observed effect of the lysozyme amount deposited at the target, on the proton/ cation distribution, for a given lysozyme charge state in DESI (Figure 1a) compared that obtained in ESI (Figure S1). Despite the nascent offspring droplet heterogeneity, their initial lysozyme concentration contributes essentially to the CSD as well as to the distribution of the proton/metallic cation ratio. The signal “zooms” for the 8+ lysozyme species, around m/z 1780-m/z 1800 (Insets of Figures S1 and 1a) display the cationized [Lyso + 6H + Ca]8+ forms (i.e., LysoCa8+) rather than its isobaric [Lyso + 7H + K]8+ form (i.e., LysoK8+) (supplementary material S2), in addition to Lyso8+.
Interestingly, for each charge state, the normalized relative abundances of the Lyson+ and LysoCk n+ ions [i.e., (ILyson++ΣILysoCaq n+=100%] depend on the charge (n) (Figure S2). So, for the charge state i+ which increases from 7+ to 13+ in ESI (Figure S2a), the Lyson+ and LysoCk n+ relative abundances are respectively equal to (96 ± 3)% and (4 ± 3)%, with a 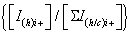 ratio (noted as Ri+) almost constant within the experimental errors.
ratio (noted as Ri+) almost constant within the experimental errors.
Consequently, in ESI, this ratio can be considered almost constant within the experimental errors. This trend is not that of the DESI experiments with methanol/water (Figure S2b). Indeed, when the charge state increases from 7+ to 11+, a significant enlargement of the Lyson+ relative abundance from 62% to 90%, and vice versa from that of LysoCk n+ which present an abundance decrease from 38% to 10%. It results in a Ri+ ratio multiplied by a factor of # 5.5. Furthermore, the larger contribution of cationized multi-protonated Lysozyme is clearly illustrated by deconvolution of the DESI mass spectrum of Figure 1a, which indicates that: (i) approximately 8% of ions are cationized, and (ii) the cationization is reinforced for the lower charged species. From these features, it appears a significant variation of the Z(h)av and Z(h/c) av values (calculated from Zav=9.0) which are 9.1 and 7.9, respectively. This behavior is consistent with different studies which enlightened this reinforced cationization of the proteins in DESI [4,36].
In the ESI experiments, independently of the maximum number of present protons on the micro-droplet surface, the fast exchanges of metallic ion/proton taking place in solution (or into the charged aggregates) can explain the almost constant cationization, maintained at low level. This yields formation of strongly charged lysozyme/ solvent aggregates, which are then desolvated under reduced pressure conditions. On the other hand, this means that, in ESI, the residual alkaline/alkaline earth ions (naturally present in the native proteins) are almost completely exchanged by protons in droplet/aggregate systems, leading to reduction of the metallic ion contribution into the naked multi-charged proteins. This behavior cannot occur in the DESI mode since the offspring droplets (emitted from the thin layer on the bulk sample onto the target) [43] are unlikely saturated by a lot of protons. Consequently, in such secondary droplets, the number of protons is not enough large [113,114] to allow an almost complete displacement of all alkaline/alkaline earth ions and to provide cation-free- multiprotonated lysozyme. Thus, after desolvation of the desorbed multicharged aggregates, residual metallic cations can be carried by the multi-protonated lysozyme. This explanation is consistent with the droplet pickup DESI mechanism model [4], which considers that secondary microdroplets are significantly smaller than those formed in ESI [43,64,116], and thus, should very likely carry less protons (vide supra). Consequently, to achieve the final ionization, cation/proton exchanges are significantly larger in ESI than in DESI from the charged aggregates (Supplementary material S2, S3).
Effects of the sample dilution in protic solvent on the charge state distribution, and on the extent of proton/cation exchange
For this purpose, we studied the evolution of the CSD and its corresponding Zav relative values depending on the amount of lysozyme deposited on the PTFE target of DESI experiments (Figures S3 and 2a). When the lysozyme amount decreased from 50 pmoles to 0.05 pmol, it appears: (i) a charge state shifting towards higher values up to 12+ (Figure S3) and (ii) a monotonic increase of Zav from 8.5 to 10.2 (Figure 2a). Such behavior does not differ strongly from that observed in the ESI mode [81-83,88-91,117]. However, the ESI signal is maintained constant in time, due to continuous droplet renewal, contrasting to that occurred from the DESI experiments which involve three periods. Indeed, due to the lysozyme solubility in water/methanol mixture, we can consider that the lysozyme concentration (i) reaches its maximum as soon as the first acetone layers are deposited on the target, (ii) remains constant during the period of the continuous consumption, and (iii) decreases rapidly when the sample bulk disappears. Furthermore, the average size and net charge distribution of offspring droplets should be approximately constant in time because of the momentum transfer from the multiple impacting primary droplets of solvent mixture with a constant composition (the layer surface being constantly renewed). Consequently, under these conditions, the charge number on the emerging droplets remains almost independent of analyte concentration; the decrease of the deposited lysozyme amount should result in a Zav increasing. This is consistent with the monotone and slight lowering Zav evolution observed by increasing the deposited lysozyme amount on the PTFE target (Figure 2a).

Figure 2: Effects of lysozyme deposit amount on the target submitted to the primary droplet beam prepared from 0.1% formic acid in methanol/water: 8/2 as sprayed solvent on evolution of (a) the charge state average Zav of multiply charged lysozyme versus logarithmic lysozyme deposit amount scale and (b) the abundances (relative to 100%) of the height-charged massif (represented by bars) of Lyso8+ ( ) and SLysoCk8+ (
) and SLysoCk8+ (  ) versus the lysozyme deposit amount.
) versus the lysozyme deposit amount.
In addition, this explanation is also consistent with the increased contribution of cationized molecules with higher deposited lysozyme amount (Figure 2b), which results in a higher proton consumption. Indeed, e.g., the relative Lyso8+ abundance decreases while that of the cationized LysoCaq 8+ forms (q equal to 1 or 2) increases as the lysozyme deposit enhances from 0.050 pmol to 50 pmoles as shown in Figure 2b. This effect is consistent with the interpretation which considers that an increase in native lysozyme deposited (in the form of salts) leads to a less proton/metal cation exchanged in the layer and offspring droplets, and therefore, reinforces the relative contribution of cationized multi-protonated LysoCaq n+ forms of lysozyme (Figure 2b). This interpretation supports the observed effects of the deposited lysozyme amount on the CSD, and the proton/cation distribution for a given charge state carried out by lysozyme in the DESI mass spectrum (Figure 2b). Conversely, it is possible to apply the latter particular effect to explore qualitatively the evolution of lysozyme concentration into the offspring droplets when an aprotic solvent such as acetone is sprayed in DESI experiments.
Unusual lysozyme ionization from primary droplet beam of aprotic solvent as anhydrous acetone
Replacement of the protic spray solvent (i.e., methanol) by an aprotic one, as acetonitrile, led to a dramatic suppression of the lysozyme signal by more than two orders of magnitude in DESI mode. This behavior was somewhat unexpected because in ESI experiments, acetonitrile [42,52] is currently used as a very effective solvent for lysozyme ionization (no more discussion herein) in DESI. This degradation of the signal is probably due to a slower solubilization than that achieved with a mixture of methanol-water. However, this possibility is not confirmed with the anhydrous acetone use as shown in Figure 1b, since an important signal appears, even if solubilization of lysozyme is less effective than with acetonitrile. Indeed, acetone presents very weak solubilization efficiency for proteins and rather favors their aggregation and, their precipitation [118]. The multiplycharged lysozyme production (Figure 1b) led us to explore deeper the influence of properties of fine droplets prepared from sprayed solvent on the ionization of proteins in DESI, especially with an aprotic one such as anhydrous acetone. This should enlighten certain features about the ionization mechanism under sprayed acetone conditions. Note that acetone is not commonly used as an ESI solvent. Its mixture with water, in 50/50 or 99/1 ratios, shows desorption/ionization of ferrocene derivatives [119] and oligomeric compounds [120], respectively. As far as we know, under ambient ionization conditions, anhydrous acetone has never been successfully applied to protein analysis. From the characteristic properties of acetone, the nascent offspring droplets should contain lysozyme within a very lower concentration than that with offspring droplets provided from the sprayed methanol/water. However, due to the fast evaporation of the acetone, the size of the survivor droplets in front of the transfer capillary will be significantly reduced. That can lead either to an increase of the yield of protein ionization/desorption, or to a faster evaporation of the offspring droplets reaching the formation of their charged residue with lysozyme as aggregates characterized by both the charge and size distributions.
`With a primary sprayed acetone droplet beam, the DESI mass spectrum of 5 pmoles deposited lysozyme (Figure 1b) displays an unexpected broad CSD from Lyso7+ to Lyso12+, with Lyso10+ as main charged species and a Zav charge state average of (9.4 ± 0.3), whereas with acidified methanol/water mixture, with the same deposited amount, the Zav value slightly decreases to (9.0 ± 0.1). A priori, this moderate value seems to be inconsistent with that expected by considering both the methanol [95,96] and water [95,121] gas phase basicities (i.e., GB(CH3OH)=724.5 kJ.mol-1 and GB(H2O)=660.4 kJ.mol-1), which are significantly lower than that of acetone [GB(acetone)=789.6 kJ.mol-1)] [95,122]. Consequently, a reverse trend should be observed if charge state depends mainly on the relative GB (and PA) values as evidenced in ESI various studies [50,123-127].
The Zav variation from 9.0 to 9.4 suggests that the “equivalent lysozyme deposit” in methanol/water, into the thin target layer (or in the offspring droplets), could roughly be estimated, according to Figure 2a, to 1.41 pmole (or much less). In addition, from the spraying anhydrous acetone DESI experiments, the [LysoCaq 8+/Lyso8+] ion abundance ratio (Inset of Figure 1b) is significantly higher than that provided from experiments performed with acidified methanol/ water DESI spray (Inset of Figure 1a). Indeed, the cationized species contribution is 30% i.e., Z(h)av=9.6, and Z(h/c)av=8.8, calculated from deconvoluted acetone spray mass spectrum, significantly higher than the 8% values (i.e., Z(h)av=9.1, and Z(h/c)av=7.9) characterizing DESI mass spectrum performed with CH3OH/H2O (Figure 1a).
Consequently, because of the limitation in the charge number carried by acetone droplets, the alkaline/alkaline earth cation/proton exchanges are also limited. In this way, when using anhydrous acetone instead of methanol/water mixture, the DESI mass spectrum displays a reduction of the absolute intensity of the base peak from 8.105 a.u. (a.u. is arbitrary unit) to 9.104 a.u. (Figure 1a and 1b). This is due to the reduced available proton number relatively less numerous with anhydrous acetone, a particular aprotic solvent, than with protic solvents. This conclusion was expected because of the high volatility of solvent, which in DESI mode plays a significant effect, and thus, must be considered in the formation of multi-charged aggregates with lysozyme.
These considerations are consistent with the highest Zav values (i.e., 9.4), if it is considered that the previous deposited lysozyme equivalent of 1.41 pmole (Figure 2a) was strongly overestimated on nascent offspring droplets (or in thin surface layer). Furthermore, this analysis explains the shift of the CSD values towards higher values, despite the lower number of available protons in the nascent offspring droplets, as regards numerous present protons released by the sprayed protic solvent, as well as the incomplete exchange of the metallic cations by the available protons.
Comparison of the lysozyme ion signal duration according to the sprayed solvent in DESI
Knowing the aprotic character of sprayed anhydrous acetone and its relative low boiling point (56°C), the resulting offspring droplets will undergo faster evaporation than those generated from the 8/2 methanol/water mixture (boiling point higher than that of acetone). As it was demonstrated from the ESI process [127], one can expect a similar trend in DESI, e.g., that an elevated rate of acetone evaporation is associated with more offspring droplets bearing a high surface charge density and then, production of higher charged lysozyme ions since the lower lysozyme concentration (weak solubility in acetone). Finally, the average equivalent concentration of lysozyme (dissolved and/or adhered to the droplet surface) in the nascent offspring droplets of acetone can be roughly estimated from: (i) the amount of deposited lysozyme (~5 pmoles), (ii) the solvent flow rate (5 μL.min-1), and (iii) the average duration for a total spot desorption close to (5 ± 1) min (i.e., until the entire signal intensity extinction). Thus, the average concentration of lysozyme in the nascent acetone offspring droplets was roughly estimated to be 0.2 μM i.e., ten times less than that in methanol/water (considered as 2.1 μM, supplementary material S1) for a shorter signal duration (0.48 min for methanol/water vs. 5 min for acetone anhydrous).
The corresponding total ion current (TIC) of 1.54 × 106 a.u. (with sprayed anhydrous acetone) was calculated by the signal integration during 5 min. Comparison with DESI experiments based on the acidified spray methanol/water mixture [i.e., duration of (0.48 ± 0.2) min for a TIC value of 10.3 × 106 a.u.] leads to a significant ion abundance diminution, corroborating the key role of lysozyme dissolution (and droplet surface adhesion) in the DESI process as previously evoked [63,67,92]. This justifies the larger duration required, for a sufficient volume of primary sprayed acetone anhydrous to impact the target, and to completely dissolve the analyte, leading to offspring droplets released from the thin solvent layer [66,67].
By using the TIC values and signal durations, the relative averages of ion production rates were estimated at (21.5 ± 2).106 a.u./min and (3.1 ± 0.6).105 a.u./min, for the sprayed acidified methanol/water and dry acetone, respectively. Consequently, with the latter, the lysozyme signal is reduced by more than 69 times. This emphasizes the importance of the analyte dissolution/solvation as well as the available proton number in offspring. It is very low in dry acetone compared to acidified methanol/water mixture. Despite its inefficient solubilizing capacity, acetone anhydrous produced significant ionic signal duration in DESI process, due to its low solubility and weak sample consumption.
The acetone effectiveness on the lysozyme desorption/ionization in the DESI experiments can undoubtedly be related to the surface PTFE properties, with its strong hydrophobic character [128]. Thus, a limited sprayed solvent dispersion improves the film homogeneity on PTFE (sample/solvent amount per unit of surface). Moreover, the fast acetone evaporation supports the formation of small ionic aggregates with enough charges, although the available charge number is lower than that obtained with methanol/water mixture, leading to a more efficient desolvation at the skimmer.
Origin of protons required for multi-protonation of lysozyme in DESI acetone anhydrous
In these experiments, a relevant question appears about the origin of the protons carried by multi-protonated lysozyme. A first assumption may be based on formation of the CH3COCH3 +? molecular ions into the dry primary charged droplets. The formation of the odd-electron molecular ions is considered to take place through an electrochemical process in the sprayer. In addition to the detection of the protonated acetone, presence of its odd-electron molecular ions in the background of DESI, is mainly observed, but in very low abundance results not reported). This behavior is similar to that observed in the acetone/ESI [129-132]. If in sprayed dry acetone, this ion can, in solution, indirectly tautomerize into the less stable ionized enol (i.e., H2C=C(OH)CH3 +?) thanks to the protic solvent. Such tautomerization can occur in the gaseous phase by the formation of an adduct-ion resulting from ion-molecule reaction with neutral acetone via ”self-catalysis” [133] and through an homo-dimer linked to the radical ion by hydrogen bonds [133-136]. This dimeric species, as the drawing force, orients dissociation towards the CH2=C(O?)CH3 radical release [137,138]. Consequently the (CH3)2C=OH + formation could be the origin of the lysozyme multi-protonation process.
To examine this hypothetical pathway, d6-acetone (i.e., CD3COCD3) was used as solvent spray. Surprisingly, three interesting features can be underlined from the mass spectrum (Supplementary material, Figure S4a) of lysozyme and its deconvolution (Supplementary material, Figure S4b): (i) detection of multi-protonated lysozyme without incorporation of several deuterons. Indeed, the lysozyme average molecular mass provided from the sprayed light anhydrous acetone (i.e., Mwave,exp=14305.20 u, vide infra) compared to that obtained from deconvoluted DESI/d6-acetone mass spectrum (i.e., Mwave,exp=14305.9183 u values) displays a shift of less than one u on the average molecular mass; (ii) a spectacular shifting of the average of the charge state Zav, decreasing from 9.4 (Figure 1b) to 6.7 (i.e., Z(h,d) av=7.0, and Z(h,d/c)av=6.6 (Figure S4a), although the presence of only one deuteron at maximum, (vide infra); and finally, (iii) an increase of the cation contribution (calculated from deconvoluted mass spectra) (Figure S4b), and enlarged from 30% to 70% for light and heavy sprayed acetone, respectively.
The lowering of the Zav value in (ii) (i.e., a shift of the charge state distribution of multi-protonated lysozyme from Z(h)av=9.6 to Z(h,d) av=7.0) must reflect a decrease of the available proton number when d6- acetone spray is used. This interpretation is consistent with the increase of the cationized form contribution from 30% to 70% in (iii), since the available proton number is significantly weakened. In addition, as for Z(h,d) av lower than Z(h)av, Z(h/c)av (i.e., 8.8) is decreased to Z(h,d/c)av=6.6. More importantly is the incorporation of almost 9 protons for Lyso9+ in (i) rather than the expected nine deuterons. This restriction could be explained by fast H/D exchanges from labeled aggregates occurring in the gaseous phase and/or in the surrounding wet environment of target.
In order to explore this possible H/D back stepwise exchange pathway in gaseous phase experiments with sprayed anhydrous CD3OD solvent (without D2O) were performed instead of the sprayed CD3COCD3 use. Indeed, the presence of heavy water may prevent the detection of back D/H exchange that could be a priori possible with the ambient humidity. In fact, a broad H/D exchange distribution is observed with, e.g., an average of 48 deuterons introduced in the Lyso9+ ion, 9D+ for ionization and 39 for H/D exchange (non-reported data). This shows that if the direct H/D exchanges take place, back reactions with the atmospheric ambient humidity do not occur. Thus, such D/H back exchanges must be ruled out to explain the quasi-absence of the labeled multi-charged lysozyme with the (d6) labeled acetone spray. On the other hand, this confirms that the above mechanism, involving possible formation of protonated acetone (or deuterated d6-acetone) does not take place in these conditions, and is not directly responsible for multiple-proton transfers to lysozyme.
Interestingly, the spraying of labeled acetone implicates a significant decrease of available protons, as shown by both the Zav value and H+/ Ca2+ exchange reduction (vide infra) i.e., (ii) and (iii)). The decrease of the deuteron/proton number can be attributed to primary isotopic effects which slow down the formation of the deuterated species of lysozyme via multi-deuteron/proton transfers. Consequently, one may ask: what is (are) the entity(ies), responsible for the formation of multiprotonated lysozyme (Lyson+) and its cationized counter-part, observed in DESI with sprayed dry acetone?
Let us to recall that in APPI, acetone is known as a doping agent to assist APPI, because otherwise photon-energy (~10 eV) of Kr VUV lamp [139,140] is insufficient to ionize usual protic solvents (e.g., water and methanol having high ionization energy). Doping participate to solvent protonation (e.g., IEwater:12.62 eV), presumably by exothermic stepwise consecutive collisions on acetone molecular ion with at least 2H2O (-63 kJ.mol-1) as reported in Equation 4 [95,96,141].
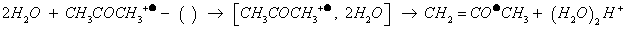 (4)
(4)
However, only the production of mono-protonated molecules in gas phase would take place (due to same charge polarity repulsion in the ion-ion interaction) in contrast to that observed in DESI, because of multi-protonated forms with sprayed dry acetone. Thus, APPI-like process cannot be considered. Despite the process taking place in APPI, the ambient water trace role could be finely observed in DESI process even with a spray of anhydrous acetone. Such atmospheric pressure conditions do not prevent from the surrounding moisture, which could be the source of protons responsible for the multi-protonation processes in DESI when anhydrous acetone is sprayed. Note that in the APCI, protonation of acetone does not occur when a dry compressed gas (e.g., D-nitrogen) is used, whereas with the T-air gas, protonation of acetone appears [142], likely similar adsorption of the ambient water takes place with singly charged small aggregates.
In order to check this assumption, three different experiments based on sprayed anhydrous acetone, were performed under rarefied air conditions on the target of deposited lysozyme, by introducing postflow argon constituted by: (i) dry argon flushing the closely surrounded target. It causes a total extinction of the lysozyme signal (Figure 3a). Taking into account the very low GB of argon, (GBAr=345.8 kJ.mol-1, estimated from proton affinity) [143] the proton transfer from ambient multi-protonated lysozyme to Ar is thus excluded. Then, a question arises, why dry argon led to such an ionic signal removal? (ii) preliminary humidified argon stream (Ar/H2O, Figure 3b). The signal is restored with reduction of the charge state average from (9.4 ± 0.3) (Figure 1b) to (8.5 ± 0.3), with Z(h)av=8.7 and Z(h/c)av=8.1, although the argon flow was moistened prior to. This experiment strongly supports the ambient humidity role in DESI source environment in lysozyme multiprotonation. Indeed, the dilution of the water vapor into argon (larger dilution than that in the ambient atmosphere) results in the decrease of the previous Zav values. Thus, the protons would originate mainly from ambient humidity rather than directly from the sprayed anhydrous acetone as shown from the d6-acetone experiments (vide supra); (iii) labeled Ar/D2O post-flow (Figure 3c) to confirm the ambient water role. The mass spectrum exhibited a narrow CSD from 11+ to 5+ related to a Zav decrease from (8.5 ± 0.3) with Ar/H2O stream to (7.7 ± 0.2) with Ar/D2O. Furthermore, it appears a large deuteron incorporation confirming the role of ambient water (or heavy water) for lysozyme multi-protonation (or multi-deuteration). A deconvoluted DESI/dry acetone mass spectrum, with Ar/D2O post-flow, displays an increase of the metallic ion contribution (alkali/alkaline earth cations) as 43% compared to 35% observed in the unlabelling post-flow experiments. On the other hand, the Z(h)av and Z(h/c)av terms equal to 8.7 and 8.1, with the Ar/H2O experiment, respectively decrease to Z(h,d)av=7.9 and Z(h,d/c) av=7.4, under the labeling post flow conditions. This trend is consistent with a lowering of the available proton/deuteron number with the surrounding D2O, very likely, due to the previous considered isotopic effect during proton/deuteron transfers (and/or exchanges).

Figure 3: DESI mass spectra of 5 pmole lysozyme deposited on target and desorbed using acetone spray under post flow conditions: (a) signal extinction with Ar as ambient gas, (b) with argon preliminary bubbled in light water, and (c) with Ar preliminary bubbled in D2O.
For each charge state, the natural isotopic clusters are shifted to higher m/z ratios and the isotopic pattern distribution is broader than that corresponding to the natural one (Figure 4). For instance, the estimated centroid of the non-cationized Lyso9+ ion shifted from m/z 1590.5044 to m/z 1591.3182 by 0,8138 (i.e., 7.3241 u, corresponding on average to introduction of more than 7 deuterons, Figure 4). Furthermore, the isotopic signal distribution width, measured at 10% of isotopic pattern height, presents a peak number increasing from 12 (natural isotopic distribution with Ar/H2O, Figure 3b) to 24 (distribution enlargement with Ar/D2O, Figure 3c) i.e., a maximum of 12 deuterons, far from completion, although several mobile protons were exchanged in addition to the 9D+ charging Lyso9+. Since deuterons are not directly supplied from d6-acetone (see above), the shift of the isotopic clusters observed with post-Ar/D2O flow (Figure 4) allows to consider the main part of the labeled surrounding water for adding 9D+ (plus the H/D exchanges). These clearly indicate that the multi-protonated lysozyme, arising from the multi-step post-spray, is promoted by the air moisture.

Figure 4: Superimposition of lysozyme 9+ charge state (Lyso9+) isotopic cluster distribution from the DESI experiments using sprayed light acetone using post-flow argon preliminary bubbled in light water ( - - - ) and heavier D2O in ( —— ). Comparison of the isotopic cluster distributions for estimating contribution of deuteron isotopes to lysozyme multi-protonation showing a shift by D=0.81 m/z of the broad isotopic distribution centroid of the Lyso9+ ion.
Under Ar/D2O post-flow conditions, the overall duration of both the target layer and offspring droplet production, can be estimated about 10 to 100 μsec. It is enough to introduce on average D+ and 12 at the maximum (i.e., 9D+ and 3 H/D exchanges) and to form labeled Lyso9+ ions. Several studies [91,144] on the gas-phase H/D exchanges in ESI with multi-protonated lysozyme were performed by ion storage. After one-second storage of the Lyso9+ ion under labeling gas phase conditions, approximately 60 H/D exchanges were introduced [144]. This result is comparable to that obtained after 10 s of the Lyso9+ ion storage, since 63 D are introduced from gas phase H/D exchanges [145]. Without ion storage, the Lyso9+ ions yield only 26 H/D exchanges, during ion accumulation and analysis cycles, corresponding to a few hundreds of milliseconds in the labeled gas phase environment [145]. As noted from the DESI/CD3OD experiments (vide supra), 48 D were introduced into the Lyso9+ ion. Those corresponded to a similar gas phase exchange of Lyso9+ provided with same labeled reagent, after ion storage of 3-4 sec into an ion trap [145]. This means a larger ion reactivity takes place in DESI as enlightened using nucleophilic reagent in DESI [8,27,69,144-146].
In DESI, it can be considered from the previous results, that prompt adsorption of ambient water traces (or heavy water) on charged droplets or/and the impacted surface layer may lead to primary charged species hydration (i.e., the odd-electron acetone ions). Similar reactions, described by Momoh [136], can also occur on the charged surface (or on droplets) subjected to the atmospheric moisture. It results in the provided aqueous layer (or micro-droplets) of acetone on sample surface enhancing lysozyme extraction and thus, the ion abundance increase. Consequently, in the Ar/D2O post-flow experiments, the (H3C)2CO+ molecular ions present in droplets, after reaction with adsorbed water, provide solvated protons, which are carried out, e.g., by labeled water into charged aggregates (Equation 4). Furthermore, the solvated proton/deuteron in the possible (D2O) nH+ clusters are randomized and/or exchanged by multiple collisions with D2O to give rise to formation of a formal (D2O)nD+ and (D2O)nH+ mixture. The result is an accumulation of protons/deuterons either at the surface of droplets, or at the target thin layer to produce charged aggregates which give rise, after stepwise desolvation, to formation of multi-deuterated/protonated lysozyme species.
Finally, unlike reactions of charged aggregates through ionmolecule reactions in gaseous phase, macroscopic processes could be considered, especially those implicating multi-solvated systems constituted by charged aggregates of acetone/protein with water provided by the atmospheric moisture. It may result in charged aggregates of water-acetone-lysozyme, odd-electron acetone ion enolisation assisted by water, which yields in the protein protonation. Similar mechanism was described for small size ions, in higher vacuum experiment of a FTICR instrument. Indeed, assisted radical-ion isomerization yielding distonic ions [134-137] from short life-time adduct ions were achieved with trace of water.
However, if such a mechanism explains the proton origin, it cannot rationalize multiple-proton transfers to lysozyme from the ion-molecule processes (vide supra). Only by considering that, after the adsorption of water molecules on the primary droplets, and more likely in the liquid layer wetting the target, where the charges are accumulated, the required solvated proton formation (formed as described above) occurs. The role of water is even more enhanced than the evaporation of the acetone is faster than that of water, so that the water enrichment occurs in offspring droplets when these ones approach the step of the charged aggregate emergence. This leads directly (or via offspring droplets) to fast desorption of the multi-protonated lysozyme aggregates or fast production of solvated and charged residues which are desolvated at the reduced pressure skimmer zone. Thus, this means that the multiprotonated aggregates can be first formed from the offspring droplets, and then, released from the wet surface layer on the target. This implies that a significant number of protons are already available in the wet layer on the target. On the other hand, this means that water molecules are rapidly adsorbed in primary droplets (or at the target layer) to react with ionized acetone and give protonated reagent. It results a large number of protons yielding a sufficient number of protonated water molecules to produce the multi-protonated lysozyme from desorbed charged aggregates.
Conclusions
The DESI potentiality to produce large size ions in gas phase is important, since the use of aprotic solvents is possible, even if they do not directly provided protons for ionization. Thus, proteins can be multi-protonated by inappropriate solvents such as aprotic solvent, e.g., anhydrous acetone. An unusual origin of protonated agent seems arise for generating the multi-protonated molecules in the case of deposited lysozyme on target within a long duration. Indeed, when using dry d6- acetone spray, Lyso9+ does not incorporate more than one deuteron over the nine expectable, whereas with sprayed CD3OD, 48 deuterons (nine ionizing deuterons and H/D exchanges) are incorporated thanks to the direct in situ CD3OD2 + formation (from the initial CD3OD+? ion reacting with the CD3OD neutrals). Such as sequence is hindered with odd-electron acetone because its reaction with neutral acetone does not provide protonation. This indicates that from sprayed dry acetone, ionizing solvated protons are not directly generated. This abnormal behavior was interpreted by considering the role of the ambient moisture as the cause of the lysozyme multi-protonation. This takes place through: (i) condensed phase by adsorption of the water molecules on the micro-droplets (or/and at the thin bulk surface layer), or/and (ii) the macromolecular systems as multi-charged aggregates submitted to multiple solvations by thermal collision cascades with the ambient water molecules, where the solvated D+ agent is accumulated.
Such indirect reactions, via the odd-electron acetone ion into the charged complex aggregates, give rise to formation of a lot of available solvated protons (or deuterons) yielding intra-aggregate proton (or deuteron) transfers for the multi-protonation (or deuteration) of lysozyme. Otherwise, convincing experiments give evidence such a process.
They are based on the dry argon post-spray flow introduction which leads to the lysozyme ion suppression by rarefying the moisture around the target. Reversely, the wet argon experiment results in the recovery of the multi-protonated lysozyme signals which, by using post-flow of Ar/D2O, is shifted due to the multi-deuteron incorporation (i.e., an average of 7D with a maximum of 12D for Lyso9+). Note, that no large gas phase D/H back exchange takes place as evidenced by introducing irreversibly 48D, using protic CD3OD solvent spray. On the other hand, preservation of cationization is shown as depending upon the available protons/deuterons on the thin target layer or/and on the offspring droplets. The average charge state of cationized multi-protonated lysozyme is useful as probe for available proton comparison in function of sprayed solvent and experimental conditions. All these results evidence that solvated H+/D+ found their origin from light (heavy) water from ambient humidity rather than directly from sprayed anhydrous acetone in DESI. From the sprayed protic solvent (here, methanol or water), the ambient water traces is not needed since solvated protons are directly produced in the primary spray (or in the target layer). Most likely, the offspring droplets carried out enough protons to promote production of the multi-protonated aggregates. Finally, if acetone is a wrong solvent for proteins, it allows indirectly multi-protonation steps with a large charge distribution and a long duration of the signal.
Acknowledgements
Authors Anna Warnet and Nicolas Auzeil contributed equally to this work. Thanks to UPMC and CNRS and the SM3P platform where analyzes were conducted. Thanks to Antony Mallet, for his advices, his precious help and his fruitful discussions.
References
- Takáts Z, Wiseman JM, Gologan B, Cooks RG (2004) Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 306: 471-473.
- Venter A, Nefliu M, Cooks RG (2008) Ambient desorption ionization mass spectrometry. Trends Anal Chem 27: 284-290.
- Cotte-Rodríguez I, Mulligan CC, Cooks RG (2007) Non-proximate detection of small and large molecules by desorption electrospray ionization and desorption atmospheric pressure chemical ionization mass spectrometry: instrumentation and applications in forensics, chemistry, and biology. Anal Chem 79: 7069-7077.
- Takats Z, Wiseman JM, Cooks RG (2005) Ambient mass spectrometry using desorption electrospray ionization (DESI): instrumentation, mechanisms and applications in forensics, chemistry, and biology. J Mass Spectrom 40: 1261-1275.
- Cooks RG, Ouyang Z, Takats Z, Wiseman JM (2006) Ambient Mass Spectrometry. Science 311: 1566-1570.
- Cotte-Rodríguez I, Takáts Z, Talaty N, Chen H, Cooks RG (2005) Desorption electrospray ionization of explosives on surfaces: sensitivity and selectivity enhancement by reactive desorption electrospray ionization. Anal Chem 77: 6755-6764.
- Badu-Tawiah AK, Eberlin LS, Ouyang Z, Cooks RG (2013) Chemical aspects of the extractive methods of ambient ionization mass spectrometry. Annu Rev Phys Chem 64: 481-505.
- Haddad R, Sparrapan R, Eberlin MN (2006) Desorption sonic spray ionization for (high) voltage-free ambient mass spectrometry. Rapid Commun Mass Spectrom 20: 2901-2905.
- Dixon RB, Sampson JS, Muddiman DC (2009) Generation of multiply charged peptides and proteins by radio frequency acoustic desorption and ionization for mass spectrometric detection. J Am Soc Mass Spectrom 20: 597-600.
- Haapala M, Pol J, Saarela V, Arvola V, Kotiaho T, et al. (2007) Desorption atmospheric pressure photoionization. Anal Chem 79: 7867-7872.
- Heron SR, Wilson R, Shaffer SA, GoodlettDR, Cooper JM (2010) Surface Acoustic Wave Nebulization of Peptides as a MicrofluidicInterface for Mass Spectrometry. Anal Chem 82: 3985-3989.
- Chen TY, Lin JY, Chen JY, Chen YC (2010) Ultrasonication-assisted spray ionization mass spectrometry for theanalysis of biomolecules in solution. J Am Soc Mass Spectrom 21: 1547-1553.
- Nemes P, Vertes A (2007) Laser ablation electrospray ionization for atmospheric pressure, in vivo, and imaging mass spectrometry. Anal Chem 79: 8098-8106.
- Cody RB, Laramée JA, Durst HD (2005) Versatile new ion source for the analysis of materials in open air under ambient conditions. Anal Chem 77: 2297-2302.
- McEwen CN, McKay RG, Larsen BS (2005) Analysis of solids, liquids, and biological tissues using solids probe introduction at atmospheric pressure on commercial LC/MS instruments. Anal Chem 77: 7826-7831.
- Wu C, Dill AL, Eberlin LS, Cooks RG, Ifa DR (2013) Mass spectrometry imaging under ambient conditions. Mass Spectrom Rev 32: 218-243.
- Ma X, Zhang S, Zhang X (2012) An instrumentation perspective on reaction monitoring by ambient mass spectrometry. Trends Anal Chem 35: 50-66.
- Chipuk JE, Brodbelt JS (2008) Transmission mode desorption electrospray ionization. J Am Soc Mass Spectrom 19: 1612-1620.
- Espy RD, Badu-Tawiah A, Cooks RG (2011) Analysis and modification on surfaces using molecular ions in the ambient environment. Curr Opin Chem Biol 15: 741-747.
- Peters KC, Comi TJ, Perry RH (2015) Multistage Reactive Transmission-Mode Desorption Electrospray Ionization Mass Spectrometry. J Am Soc Mass Spectrom 26: 1494-1501.
- Liu P, Zheng Q, Dewald HD, Zhou R, Chen H (2015) The study of electrochemistry with ambient mass spectrometry. Trends Anal Chem 70: 20-30.
- Lu M, Liu Y, Helmy R, Martin GE, Dewald HD et al. (2015) On line Investigation of Aqueous-Phase Electrochemical Reactions by Desorption Electrospray Ionization Mass Spectrometry. J Am Soc Mass Spectrom 26: 1676-1685.
- Justes DR, Talaty N, Cotte-Rodriguez I, Cooks RG (2007) Detection of explosives on skin using ambient ionization mass spectrometry. Chem Commun (Camb): 2142-2144.
- Morelato M, Beavis A, Kirkbride P, Roux C (2013) Forensic applications of desorption electrospray ionisation mass spectrometry (DESI-MS). Forensic Sci Int 226: 10-21.
- D’Agostino PA, Chenier CL, Hancock JR, Lepage CRJ (2007) Desorption electrospray ionisation mass spectrometric analysis of chemical warfare agents from solid phase microextraction fibers. Rapid Commun Mass Spectrom 21: 543-549.
- Hagan NA, Cornish TJ, Pilato RS, Van Houten KA, Antoine MD et al. (2008) Detection and identification of immobilized low-volatility organophosphates by desorption ionization mass spectrometry. Int J Mass Spectrom 278: 158-165.
- Song Y, Cooks RG (2007) Reactive desorption electrospray ionization for selective detection of the hydrolysis products of phosphonate esters. J Mass Spectrom 42: 1086-1092.
- Wells JM, Roth MJ, Keil AD, Grossenbacher JW, Justes DR, et al. (2008) Implementation of DART and DESI ionization on a fieldable mass spectrometer. J Am Soc Mass Spectrom 19: 1419-1424.
- Nielen MWF, Hooijerink H, Zomer P, Mol JGJ (2011) Desorption electrospray ionization mass spectrometry in the analysis of chemical food contaminants. Trends Anal Chem 30: 165-180.
- García-Reyes JF, Jackson AU, Molina-Díaz A, Cooks RG (2009) Desorption electrospray ionization mass spectrometry for trace analysis of agrochemicals in food. Anal Chem 81: 820-829.
- Schurek J, Vaclavik L, Hooijerink H, Lacina O, Poustka J (2008) Control of Strobilurin Fungicides in Wheat Using Direct Analysis in Real Time Accurate Time-of-Flight and Desorption Electrospray Ionization Linear Ion Trap Mass Spectrometry. Anal Chem 80: 9567 -9575.
- Mulligan CC, MacMillan DK, Noll RJ, Cooks RG (2007) Fast analysis of high-energy compounds and agricultural chemicals in water with desorption electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom 21: 3729-3736.
- Jackson AU, Werner SR, Talaty N, Song Y, Campbell K, et al. (2008) Targeted metabolomic analysis of Escherichia coli by desorption electrospray ionization and extractive electrospray ionization mass spectrometry. Anal Biochem 375: 272-281.
- Williams JP, Scrivens JH (2008) Coupling desorption electrospray ionisation and neutral desorption/extractive electrospray ionisation with a travelling-wave based ion mobility mass spectrometer for the analysis of drugs. Rapid Commun Mass Spectrom 22: 187-196.
- Kauppila TJ, Talaty N, Kuuranne T, Kotiaho T, Kostiainen R, et al. (2007) Rapid analysis of metabolites and drugs of abuse from urine samples by desorption electrospray ionization-mass spectrometry. Analyst 132: 868-875.
- Siebenhaar M, Kuellmer K, Fernandes NM, Huellen V, Hopf C (2015) Personalized monitoring of therapeutic salicylic acid in dried blood spots using a three-layer setup and desorption electrospray ionization mass spectrometry. Anal Bioanal Chem 407: 7229-7238.
- Bailey MJ, Bradshaw R, Francese S, Salter TL, Costa C, et al. (2015) Rapid detection of cocaine, benzoylecgonine and methylecgonine in fingerprints using surface mass spectrometry. Analyst 140: 6254-6259.
- Liu J, Kennedy JH, Ronk M, Marghitoiu L, Lee H, et al. (2014) Ambient analysis of leachable compounds from single-use bioreactors with desorption electrospray ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 28: 2285-2291.
- Stojanovska N, Tahtouh M, Kelly T, Beavis A, Fu S (2015) Qualitative analysis of seized cocaine samples using desorption electrospray ionization- mass spectrometry (DESI-MS). Drug Test Anal 7: 393-400.
- Manicke NE, Nefliu M, Wu C, Woods JW, Reiser V, et al. (2009) Imaging of lipids in atheroma by desorption electrospray ionization mass spectrometry. Anal Chem 81: 8702-8707.
- Bereman MS, Williams TI, Muddiman DC (2007) Carbohydrate analysis by desorption electrospray ionization fourier transform ion cyclotron resonance mass spectrometry. Anal Chem 79: 8812-8815.
- Shin YS, Drolet B, Mayer R, Dolence K, Basile F (2007) Desorption electrospray ionization-mass spectrometry of proteins. Anal Chem 79: 3514-3518.
- Myung S, Wiseman J, Valentine S, Takats Z, Cooks RG, et al. (2006) Coupling Desorption Electrospray Ionization with Ion Mobility/Mass Spectrometry for Analysis of Protein Structure: Evidence for Desorption of Folded and Denatured States. J Phys Chem B 110: 5045-5051.
- Miao Z, Chen H (2009) Direct analysis of liquid samples by desorption electrospray ionization-mass spectrometry (DESI-MS). J Am Soc Mass Spectrom 20: 10-19.
- Qiu B, Luo H (2009) Desorption electrospray ionization mass spectrometry of DNA nucleobases: implications for a liquid film model. J Mass Spectrom 44: 772-779.
- Hollenhorst MI, Lips KS, Wolff M, Wess J, Gerbig S, et al. (2012) Luminal cholinergic signalling in airway lining fluid: a novel mechanism for activating chloride secretion via Ca+-dependent Cl- and K+ channels. Br J Pharmacol 166: 1388-1402.
- Wiseman JM, Evans CA, Bowen CL, Kennedy JH (2010) Direct analysis of dried blood spots utilizing desorption electrospray ionization (DESI) mass spectrometry. Analyst 135: 720-725.
- Wiseman JM, Ifa DR, Zhu Y, Kissinger CB, Manicke NE, et al. (2008) Desorption electrospray ionization mass spectrometry: Imaging drugs and metabolites in tissues. Proc Natl Acad Sci USA 105: 18120-18125.
- Song Y, Talaty N, Tao WA, Pan Z, Cooks RG (2007) Rapid ambient mass spectrometric profiling of intact, untreated bacteria using desorption electrospray ionization. Chem Commun (Camb): 61-63.
- Montowska M, Rao W, Alexander MR, Tucker GA, Barrett DA (2014) Tryptic digestion coupled with ambient desorption electrospray ionization and liquid extraction surface analysis mass spectrometry enabling identification of skeletal muscle proteins in mixtures and distinguishing between beef, pork, horse, chicken, and turkey meat. Anal Chem 86: 4479-4487.
- Dill AL, Eberlin LS, Zheng C, Costa AB, Ifa DR, et al. (2010) Multivariate statistical differentiation of renal cell carcinomas based on lipidomic analysis by ambient ionization imaging mass spectrometry. Anal Bioanal Chem 398: 2969-2978.
- Bereman MS, Nyadong L, Fernandez FM, Muddiman DC (2006) Direct high-resolution peptide and protein analysis by desorption electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun Mass Spectrom 20: 3409-3411.
- Yang SH, Wijeratne AB, Li L, Edwards BL, Schug KA (2011) Manipulation of Protein Charge States through Continuous Flow-Extractive Desorption Electrospray Ionization: A New Ambient Ionization Technique. Anal Chem 83: 643-647.
- Ferguson CN, Benchaar SA, Miao Z, Loo JA, Chen H (2011) Direct ionization of large proteins and protein complexes by desorption electrospray ionization-mass spectrometry. Anal Chem 83: 6468-6473.
- Douglass KA, Venter AR (2013) Protein analysis by desorption electrospray ionization mass spectrometry and related methods. J Mass Spectrom 48: 553-560.
- Rao W, Celiz AD, Scurr DJ, Alexander MR, Barrett DA (2013) Ambient DESI and LESA-MS analysis of proteins adsorbed to a biomaterial surface using in-situ surface tryptic digestion. J Am Soc Mass Spectrom 24: 1927-1936.
- Przybylski C, Gonnet F, Hersant Y, Bonnaffé D, Lortat-Jacob H, et al. (2010) Desorption electrospray ionization mass spectrometry of glycosaminoglycans and their protein noncovalent complex. Anal Chem 82: 9225-9233.
- Moore BN, Hamdy O, Julian RR (2012) Protein structure evolution in liquid DESI as revealed by selective noncovalent adduct protein probing. Int J Mass Spectrom 330-332: 220-225.
- Yao C, Na N, Huang L, He D, Ouyang J (2013) High-throughput detection of drugs binding to proteins using desorption electrospray ionization mass spectrometry. Anal Chim Acta 794: 60-66.
- Liu P, Zhang J, Ferguson CN, Chen H, Loo JA (2013) Measuring Protein-Ligand Interactions Using Liquid Sample Desorption Electrospray Ionization Mass Spectrometry. Anal Chem 85: 11966-11972.
- Yao C, Wang T, Zhang B, He D, Na N et al. (2015) Screening of the Binding of Small Molecules to Proteins by Desorption Electrospray Ionization Mass Spectrometry Combined with Protein Microarray. J Am Soc Mass Spectrom 26: 1950-1958.
- Yao Y, Shams-Ud-Doha K, Daneshfar R, Kitova DR, Klassen JS (2015) Quantifying Protein-Carbohydrate Interactions Using Liquid Sample Desorption Electrospray Ionization Mass Spectrometry. J Am Soc Mass Spectrom 26: 98-106.
- Costa AB, Cooks RG (2008) Simulated splashes: Elucidating the mechanism of desorption electrospray ionization mass spectrometry. Chem Phys Lett 464: 1-8.
- Volný M, Venter A, Smith SA, Pazzi M, Cooks RG (2008) Surface effects and electrochemical cell capacitance in desorption electrospray ionization. Analyst 133: 525-531.
- De la Mora JF (2000) Electrospray ionization of large multiply charge species proceeds via Dole’s charge residue mechanism. Anal Chim Acta 406: 93-104.
- Badu-Tawiah A, Bland C, Campbell DI, Cooks RG (2010) Non-aqueous spray solvents and solubility effects in desorption electrospray ionization. J Am Soc Mass Spectrom 21: 572-579.
- Badu-Tawiah AK, Cooks RG (2010) Enhanced ion signals in desorption electrospray ionization using surfactant spray solutions. J Am Soc Mass Spectrom 21: 1423-1431.
- Kauppila TJ, Talaty N, Salo PK, Kotiaho T, Kostiainen R, et al. (2006) New surfaces for desorption electrospray ionization mass spectrometry: porous silicon and ultra-thin layer chromatography plates. Rapid Commun Mass Spectrom 20: 2143-2150.
- Ifa DR, Wu C, Ouyang Z, Cooks RG (2010) Desorption electrospray ionization and other ambient ionization methods: current progress and preview. Analyst 135: 669-681.
- Gao L, Li G, Cyria J, Nie Z, Cooks RG (2010) Imaging of Surface Charge and the Mechanism of Desorption Electrospray Ionization Mass Spectrometry. J Phys Chem C114: 5331-5337.
- Manicke NE, Wiseman JM, Ifa DR, Cooks RG (2008) Desorption electrospray ionization (DESI) mass spectrometry and tandem mass spectrometry (MS/MS) of phospholipids and sphingolipids: ionization, adduct formation, and fragmentation. J Am Soc Mass Spectrom 19: 531-543.
- Bereman MS, Muddiman DC (2007) Detection of attomole amounts of analyte by desorption electrospray ionization mass spectrometry (DESI-MS) determined using fluorescence spectroscopy. J Am Soc Mass Spectrom 18: 1093-1096.
- Ifa DR, Manicke NE, Rusine AL, Cooks RG (2008) Quantitative analysis of small molecules by desorption electrospray ionization mass spectrometry from polytetrafluoroethylene surfaces. Rapid Commun Mass Spectrom 22: 503-510.
- Takáts Z, Cotte-Rodriguez I, Talaty N, Chen H, Cooks RG (2005) Direct, trace level detection of explosives on ambient surfaces by desorption electrospray ionization mass spectrometry. Chem Commun (Camb): 1950-1952.
- Wang H, Manicke NE, Yang Q, Zheng L, Shi R, et al. (2011) Direct analysis of biological tissue by paper spray mass spectrometry. Anal Chem 83: 1197-1201.
- Sen AK, Nayak R, Darabi J, Knapp DR (2008) Use of nanoporous alumina surface for desorption electrospray ionization mass spectrometry in proteomic analysis. Biomed Microdevices 10: 531-538.
- Loriau M, Alves S, Churlaud F, Tabet JC (2009) Solvent Effects on the DESI mass spectra of Industrial polymers. Proceedings of the 57th ASMS conference, Philadelphia, USA.
- Van Berkel GJ, Tomkins BA, Kertesz V (2007) Thin-layer chromatography/desorption electrospray ionization mass spectrometry: investigation of goldenseal alkaloids. Anal Chem 79: 2778-2789.
- Liu Y, Miao Z, Lakshmanan R, Ogorzalek Loo RR, Loo JA, et al. (2012) Signal and Charge Enhancement for Protein Analysis by Liquid Chromatography-Mass Spectrometry with Desorption Electrospray Ionization. Int J Mass Spectrom 325-327: 161-166.
- Samalikova M, Grandori R (2003) Role of opposite charges in protein electrospray ionization mass spectrometry. J Mass Spectrom 38: 941-947.
- Wang G, Cole RG (1994) Effect of Solution Ionic Strength on Analyte Charge State Distributions in Positive and Negative Ion Electrospray Mass Spectrometry. Anal Chem 66: 3702-3708.
- Kuprowski MC, Konermann L (2007) Signal response of coexisting protein conformers in electrospray mass spectrometry. Anal Chem 79: 2499-2506.
- Samalikova M, Grandori R (2005) Testing the role of solvent surface tension in protein ionization by electrospray. J Mass Spectrom 40: 503-510.
- Thomas BR, Vekilov PG, Rosenberger F (1996) Heterogeneity determination and purification of commercial hen egg-white lysozyme. Acta Crystallogr D Biol Crystallogr 52: 776-784.
- Ries-Kautt M, Ducruix A (1997) Interferences Drawn from Physicochemical Studies of Crystallogenesis and PrecrystaUine State. Methods in Enzymology 276: 23-59.
- Kebarle P, Verkerk UH (2009) Electrospray: from ions in solution to ions in the gas phase, what we know now. Mass Spectrom Rev 28: 898-917.
- Deng L, Sun N, Kitova EN, Klassen JS (2010) Direct quantification of protein-metal ion affinities by electrospray ionization mass spectrometry. Anal Chem 82: 2170-2174.
- Gross DS, Schnier PD, Rodriguez-Cruz SE, Fagerquist CK, Williams ER (1996) Conformations and folding of lysozyme ions in vacuo. Proc Natl Acad Sci U S A 93: 3143-3148.
- Konermann L, Douglas DJ (1998) Unfolding of proteins monitored by electrospray ionization mass spectrometry: a comparison of positive and negative ion modes. J Am Soc Mass Spectrom 9: 1248-1254.
- Loo RR, Loo JA, Udseth HR, Fulton JL, Smith RD (1992) Protein structural effects in gas phase ion/molecule reactions with diethylamine. Rapid Commun Mass Spectrom 6: 159-165.
- Takamizawa A, Fujimaki S, Sunner J, Hiraoka K (2005) Denaturation of lysozyme and myoglobin in laser spray. J Am Soc Mass Spectrom 16: 860-868.
- Schnier PD, Gross DS, Williams ER (1995) On the maximum charge state and proton transfer reactivity of peptide and protein ions formed by electrospray ionization. J Am Soc Mass Spectrom 6: 1086-1097.
- Reimann CT, Sullivan PA, Axelsson J, Quist AP, Altmann S, et al. (1998) Conformation of Highly-Charged Gas-Phase Lysozyme Revealed by Energetic Surface Imprinting. J Am Chem Soc 120: 7608-7616.
- Verkerk UH, Peschhke M, Kebarle P (2003) Effect of buffer cations and of H3O+ on the charge states of native proteins. Significance to determinations of stability constants of protein complexes. J Mass Spectrom 38: 618-631.
- Hunter EP, Lias SG (1998) Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J Phys Chem Ref data 27: 413-656.
- Lias SG, Liebman JF, Levin RD (1984) Evaluated gas phase basicities and proton affinities of molecules; heats of formation of protonated molecules. J Phys Chem 13: 695-808.
- Petrie S, Javahery G, Wincel H, Bohme DK (1993) Attaching Handles to C60 2+: The Double-Derivatization of C60 2+. J Am Chem Soc 115: 629-634.
- Kaltashov IA, Fabris D, Fenselau CC (1995) Assessment of Gas Phase Basicities of Protonated Peptides by the Kinetic Method. J Phys Chem 99: 10046-10051.
- Williams ER (1996) Proton transfer reactivity of large multiply charged ions. J Mass Spectrom 31: 831-842.
- Miteva M, Demirev P, Karshikoff D (1997) Multiply-Protonated Protein Ions in the Gas Phase: Calculation of the Electrostatic Interactions between Charged Sites. J Phys Chem B 101: 9645-9650.
- Touboul D, Jecklin MC, Zenobi R (2008) Investigation of deprotonation reactions on globular and denatured proteins at atmospheric pressure by ESSI-MS. J Am Soc Mass Spectrom 19: 455-466.
- Touboul D, Jecklin MC, Zenobit R (2008) Exploring deprotonation reactions on peptides and proteins at atmospheric pressure by electro-sonic spray ionization-mass spectrometry (ESSI-MS). Chimia 62: 282-286.
- Burk P, Tammiku-Taul J, Tamp S, Sikk L, Sillar K, et al. (2009) Computational study of cesium cation interactions with neutral and anionic compounds related to soil organic matter. J Phys Chem A 113: 10734-10744.
- Ai H, Bu Y, Li P, Zhang C (2005) The regulatory roles of metal ions (M = Li, Na, K, Be, Mg, and Ca) and water molecules in stabilizing the zwitterionic form of glycine derivatives. New J Chem 29: 1540-1548.
- Dunbar RC (2000) Complexation of Na+ and K+ to Aromatic Amino Acids: A Density Functional Computational Study of Cation-p Interactions. J Phys Chem A 104: 8067-8074.
- Ryzhov V, Dunbar RC, Cerda B, Wesdemiotis C (2000) Cation-pi effects in the complexation of Na+ and K+ with Phe, Tyr, and Trp in the gas phase. J Am Soc Mass Spectrom 11: 1037-1046.
- Tsang Y, Wong CL, Wong CHS, Cheng JMK, Ma Nlet al. (2012) Proton and potassium affinities of aliphatic and N-methylated aliphatic-amino acids: Effect of alkyl chain length on relative stabilities of K+ bound zwitterionic complexes. Int J Mass Spectrom 316-318: 273-283.
- Jover J, Bosque R, Sales J (2008) A comparison of the binding affinity of the common amino acids with different metal cations. Dalton Trans : 6441-6453.
- Nemirovskiy O, Giblin DE, Gross ML (1999) Electrospray ionization mass spectrometry and hydrogen/deuterium exchange for probing the interaction of calmodulin with calcium. J Am Soc Mass Spectrom 10: 711-718.
- Malisauskas M, Zamotin V, Jass J, Noppe W, Dobson CM, et al. (2003) Amyloid protofilaments from the calcium-binding protein equine lysozyme: formation of ring and linear structures depends on pH and metal ion concentration. J Mol Biol 330: 879-890.
- Permyakov SE, Khokhlova TI, Nazipova AA, Zhadan AP, Morozova-Roche LA (2006) Calcium-Binding and Temperature Induced Transitions in Equine Lysozyme: New Insights From the pCa-Temperature Phase Diagrams. Proteins 65: 984-998.
- Morozova-Roche LA (2007) Equine lysozyme: the molecular basis of folding, self-assembly and innate amyloid toxicity. FEBS Lett 581: 2587-2592.
- Thomson BA, Iribarne JV (1979) Field induced ion evaporation from liquid surfaces at atmospheric pressure. J Chem Phys 71: 4451.
- Smith JN, Flagan RC, Beauchamp JL (2002) Droplet Evaporation and Discharge Dynamics in Electrospray Ionization. J Phys Chem A106: 9957-9967.
- Kaltashov I, Mohimen A (2005) Estimates of Protein Surface Areas in Solution by Electrospray Ionization Mass Spectrometry. Anal Chem 77: 5370-5379.
- Green FM, Salter TL, Gilmore IS, Stokes P, O'Connor G (2010) The effect of electrospray solvent composition on desorption electrospray ionisation (DESI) efficiency and spatial resolution. Analyst 135: 731-737.
- Przybylski M, Glocker MO (1996) Electrospray mass spectrometry of biomacromolecular complexes with noncovalent interactions-new analytical perspectives for supramolecular chemistry and molecular recognition processes. Angew Chem Int Ed Engl 35: 806-826.
- Green-Church KB, Nichols JJ (2008) Mass spectrometry-based proteomic analyses of contact lens deposition. Mol Vis 14: 291-297.
- Hartinger CG, Nazarov A, Chevchenko V, Arion VB, Galanski M et al. (2003) Synthesis, crystal structures, and electrospray ionisation mass spectrometry investigations of ether-and thioether-substituted ferrocenes. Dalton Trans 15: 3098-3102.
- Saf R, Schitter R, Mirtl C, Stelzer F, Hummel K (1996) Electrospray Ionization Mass Spectrometry Investigation of Oligomers Prepared by Ring-Opening Metathesis Polymerization of Methyl N-(1-Phenylethyl)-2-azabicyclo[2.2.1]hept-5-ene-3-carboxylate. Macromolecules 29: 7651-7656.
- Leito I, Koppel IA, Burk P, Tamp S, Kutsar M, et al. (2010) Gas-phase basicities around and below water revisited. J Phys Chem A 114: 10694-10699.
- Szulejko JE, Luo Z, Solouki T (2006) Simultaneous determination of analyte concentrations, gas-phase basicities, and proton transfer kinetics using gas chromatography/Fourier transform ion cyclotron resonance mass spectrometry (GC/FT-ICRMS). Int J Mass Spectrom 257: 16-26.
- Ogorzalek Loo RR, Winger BE, Smith RD (1994) Proton transfer reaction studies of multiply charged proteins in a high mass-to-charge ratio quadrupole mass spectrometer. J Am Soc Mass Spectrom 5: 1064-1071.
- Ogorzalek Loo RR, Smith RD (1994) Investigation of the gas-phase structure of electrosprayed proteins using ion-molecule reactions. J Am Soc Mass Spectrom 5: 207-220.
- Hunter AP, Severs JC, Harris FM, Games DE (1994) Proton-transfer reactions of mass-selected multiply charged ions. Rapid Commun Mass Spectrom 8: 417-422.
- Schnier PD, Price WD, Williams ER (1996) Modeling the maximum charge state of arginine-containing Peptide ions formed by electrospray ionization. J Am Soc Mass Spectrom 7: 972-976.
- Nguyen S, Fenn JB (2007) Gas-phase ions of solute species from charged droplets of solutions. Proc Natl Acad Sci U S A 104: 1111-1117.
- Pasilis SP, Kertesz V, Van Berkel GJ (2007) Surface scanning analysis of planar arrays of analytes with desorption electrospray ionization-mass spectrometry. Anal Chem 79: 5956-5962.
- Heaton J, Jones MD, Legido-Quigley C, Plumb RS, Smith NW (2011) Systematic evaluation of acetone and acetonitrile for use in hydrophilic interaction liquid chromatography coupled with electrospray ionization mass spectrometry of basic small molecules. Rapid Commun Mass Spectrom 25: 3666-3674.
- Keppel TR, Jacques ME, Weis DD (2010) The use of acetone as a substitute for acetonitrile in analysis of peptides by liquid chromatography/electrospray ionization mass spectrometry. Rapid Com. Mass Spectrom 24: 6-10.
- Fountain KJ, Xu J, Diehl DM, Morrison D (2010) Influence of stationary phase chemistry and mobile-phase composition on retention, selectivity, and MS response in hydrophilic interaction chromatography. J Sep Sci 33: 740-751.
- Trikoupis MA, Burgers PC, Ruttink PJA, Terlow JK (2002) Self-catalysis in the gasphase: enolization of the acetone radical cation. Int J Mass Spectrom 217: 97-108.
- Matsuda Y, Yamada A, Hanaue K, Mikami N, Fujii A (2010) Catalytic action of a single water molecule in a proton-migration reaction. Angew Chem Int Ed Engl 49: 4898-4901.
- Van der Rest G, Mourgues P, Fossey J, Audier HE (1997) The [+CH2OH, H2O] and [+CH2OH, 2H2O] solvated ions. Int. Mass Spectrom. Ion Process 160: 107-115.
- Trikoupis MA, Terlouw JK, Burgers PC (1998) Enolization of Gaseous Acetone Radical Cations: Catalysis by a Single Base Molecule. J Am Chem Soc 120: 12131-12132.
- Momoh PO, El-Shall MS (2008) Gas phase hydration of organic ions. Phys Chem Chem Phys 10: 4827-4834.
- Norrman K, Sølling TI, McMahon TB (2005) Isomerization of the protonated acetone dimer in the gas phase. J Mass Spectrom 40: 1076-1087.
- Abdel Azeim S, van der Rest G (2005) Thermochemical properties of the ammonia-water ionized dimer probed by ion-molecule reactions. J Phys Chem A 109: 2505-2513.
- Robb DB, Covey TR, Bruins AP (2000) Atmospheric pressure photoionization: an ionization method for liquid chromatography-mass spectrometry Anal Chem 72: 3653-3659.
- Marchi I, Rudaz S, Veuthey JL (2009) Atmospheric pressure photoionization for coupling liquid-chromatography to mass spectrometry: a review. Talanta 78: 1-18.
- Lias SG, Bartness JE, Liebman JF, Homes JL, Levin R.D et al. (1988) Gas-Phase Ion and Neutral Thermochemistry. J Phys Chem Ref data Suppl 17: 861.
- Kolakowski BM, Grossert JS, Ramaley L (2004) Studies on the positive-ion mass spectra from atmospheric pressure chemical ionization of gases and solvents used in liquid chromatography and direct liquid injection. J Am Soc Mass Spectrom 15: 311-324.
- Schul RJ, Passarella R, Upschulte BL, Keesee RG, Castleman AW (1987) Thermal energy reactions involving Ar+ monomer and dimer with N2, H2, Xe, and Kr. J Chem Phys 86: 4446-4451.
- Sparrapan R, Eberlin LS, Haddad R, Cooks RG, Eberlin MN, et al. (2006) Ambient Eberlin reactions via desorption electrospray ionization mass spectrometry. J Mass Spectrom 41: 1242-1246.
- Wu C, Ifa DR, Manicke NE, Cooks RG (2009) Rapid, direct analysis of cholesterol by charge labeling in reactive desorption electrospray ionization. Anal Chem 81: 7618-7624.
- Xu G, Chen B, Guo B, He D, Yao S (2011) Detection of intermediates for the Eschweiler-Clarke reaction by liquid-phase reactive desorption electrospray ionization mass spectrometry. Analyst 136: 2385-2390.
Relevant Topics
Recommended Journals
Article Tools
Article Usage
- Total views: 16111
- [From(publication date):
December-2015 - Aug 29, 2025] - Breakdown by view type
- HTML page views : 11382
- PDF downloads : 4729