Formulation, Characterization and Pharmacokinetic Evaluation of Telmisartan Solid Dispersions
Received: 03-Mar-2016 / Accepted Date: 08-Jun-2016 / Published Date: 15-Jun-2016 DOI: 10.4172/2329-9053.1000131
Abstract
Telmisartan (TEL), a BCS class II antihypertensive drug possesses poor aqueous solubility which results in poor bioavailability after oral administration. Solid dispersion approach has been widely and successfully used to improve the biopharmaceutical profiles of the poorly soluble drugs. Present study was an attempt to prepare and compare the solid dispersion (SD) of Telmisartan using a novel polymer soluplus® by employing spray drying and melt extrusion techniques. Results demonstrated an improvement in solubility of TEL by 44, 3.3, 11 and 99 times as compared to pure drug in water and buffers pH 1.2, 4.5 and 7.4 respectively, post preparation of solid dispersions. Further, similar improvement was observed in in vitro release studies highlighting the importance of formulating solid dispersion. DSC and PXRD study revealed the amorphous state of TEL in solid dispersion. The developed SD were found to be stable at 40°C/75% RH, 25°C/60% RH and 2-8°C in accordance to ICH guidelines. The pharmacokinetic study in rats revealed promising results which were correlated well with the in vitro dissolution and solubility study. Results indicated the usefulness of solid dispersions prepared using soluplus® by industrially viable techniques to improve not only the bioavailability of TEL but also their potential for commercialization.
Keywords: Telmisartan; Bioavailability; Melt extrusion; Spray drying; Commercialization
1745Introduction
Poor drug solubility is a major challenge confronted by formulation scientists worldwide and various approaches to overcome the problem are reported [1-4]. These include spray drying, freeze drying, size reduction, use of co-solvents, formation of cyclodextrin inclusion complexes and alteration of aqueous micro-environment [5].
Yet, another familiar approach has been preparation of solid dispersions by employing amphiphilic polymers such as cellulosics, Polyvinylpyrrolidone (PVP) and its copolymers, acrylates, and polyethylene oxide (PEO) and its copolymers, as an aid to improve solubilisation of such poorly water soluble molecules [6-8].
Solid dispersion technology is one of the most promising and extensively performed approach to improve the dissolution rate of insoluble compounds [9,10]. Ease of scalability, its conversion to solid dosage forms such as capsules, tablets, taste masking strips and implants are some of the advantages offered by solid dispersion over other approaches.
Solid dispersion is defined as a group of solid products consisting of at least two different components, generally a hydrophilic matrix and a hydrophobic drug. The matrix can be either crystalline or amorphous.
The drug can be dispersed molecularly, in amorphous particles (clusters) or in crystalline particles. But if the drug is converted to amorphous form and forms one phase system as drug polymer matrix, it can be classified as a solid solution, whereas if the drug exists as microcrystalline dispersion i.e., forms two-phase system, it is generally referred to as a solid dispersion [11-13].
First generation solid dispersions comprised of crystalline carriers like sugar and urea which yielded crystalline solid dispersions though they do not show any improvement in solubility and moreover the release of the drug is also delayed.
The second generation solid dispersion, prepared by dispersing the compound with amorphous carriers, yields solid dispersion or solution or suspension or mixture of both amorphous and crystalline form. In such solid dispersion system, the drug is in a solution form with help of another material, generally a polymer, either through melting and/or through the use of a solvent.
Third generation solid dispersion prepared using carriers having surface activity or self-emulsifying properties have the potential to achieve solubilisation and dissolution and enhanced stability of the drugs [14]. Latter include gelucire, inulin, polaxamers, polyethelene glycols, polysorbates and hydroxypropyl methyl cellulose.
Recent research on solid dispersions is focused towards the use of novel polymers, scalable manufacturing techniques and equipments and is heading towards the development of fourth generation solid dispersions.
They are expected to enhance the solubility and bioavailability of insoluble and high melting point drugs by formation of dispersion at molecular level using specially crafted polymers.
Telmisartan (TEL), a BCS Class II molecule (Figure 1), exhibits poor solubility in the range of pH 3-9 and increased solubility at alkaline pH 15-16. Present study aimed to prepare solid dispersions by spray drying and HME methods, employing using Soluplus® as the polymer, so as to enhance the solubility and hence the bioavailability of TEL. To the best of our knowledge this the first report investigating the usefulness of an amphiiphillic polymer i.e., Soluplus® for enhancing the solubility of Telmisartan by employing industrially feasible techniques like HME and spray drying.
Figure 1: Structure of telmisartan.
The prepared solid dispersions were suitably characterized for its assay (validated HPLC), shape and morphological attributes, and physical state using DSC & PXRD studies. Furthermore, in vitro release studies (using UV specifications) were performed to measure the extent of drug released in comparison to the free drug.
Stability studies were performed in accordance to the ICH guidelines at accelerated stability condition 40°C/75% RH and 25°C/60% RH. The control samples were stored at 2-8°C in closed vials. Finally, pharmacokinetic studies were performed in rats using validated LC/MS method to measure the enhancement in bioavailability post preparation of solid dispersion.
Experimental
Materials
Telmisartan (TEL) was procured as gift sample from Piramal Enterprises Limited. Soluplus® was purchased from BASF (Ludwigshafen, Germany). Ethanol and Methanol used in the experiments were of analytical grade (purity 99.99%) and were purchased from Merck (Merck, Mumbai, India).
Spray drying technique
The polymer Soluplus® and the drug were dissolved in a 2:1 solvent mixture (dichloromethane:methanol) with constant stirring to obtain a clear solution. The TEL-Soluplus® solution was spray dried employing a Nano Spray Drier, B-90 (Büchi®, Flawil, Switzerland) at aspirator rate 50-60%, nozzle diameter-7.00 mm, drying air (inlet) temperature- 40.0 ± 5°C, outlet temperature- 35.0 ± 5°C, with inside pressure of the spraying chamber maintained at 30-150 mbar.
To achieve optimum atomization performance in the dryer, a supply of air with a pressure of 1-2 bars was used. The spray dried powder was further collected in polyethylene bags, labeled and placed at temperatures ranging 2-8°C.
Melt extrusion technique
TEL, Soluplus® and plasticizer (PEG 1000) were used in ratio of 33:66:1. This blend was then fed into a co-rotating conical twin screw extruder (Pharma Mini HME, Thermo Scientific, Mumbai, India). The internal diameter of the die was about 1.5 mm, length was 24 mm, with a screw speed of 100 rpm and the residence time inside the barrel of the extruder was approximately 1-2 min.
The processing temperature was set at 265 ± 5°C, with torque maintained between 0.10-0.15 Nm. The extrudates were further cooled at room temperature, collected in labeled polyethylene bags and placed at refrigerated condition of 2-8°C.
Solubility studies
The solubility of the melt extrudates, spray-dried powder and TEL were determined in water, Hydrochloric acid buffer (pH 1.2), acetate buffer (pH 4.5) and phosphate buffer (pH 7.4).
An excess amount of TEL and solid dispersions were added in 5ml of the respective buffer and were stirred at 37°C at 100 rpm in a mechanical shaker (Jeio Tech, BS-21, Gyeonggi-do, South Korea). The amount of TEL solubilized was estimated by using HPLC (Agilent 1100, Agilent Technologies India Pvt. Ltd., Mumbai, India) at 298 nm after 48 hrs. The chromatographic conditions were same as described under estimation of TEL content. The analysis was performed in triplicate.
Estimation of TEL content
The assay of the formulation was determined by using HPLC system (Agilent 1100, Agilent Technologies India Pvt. Ltd., Mumbai, India) using Inertsil C18 reverse phase column (250 mm × 4.6 mm × 5 μm) with mobile phase A-potassium dihydrogen phosphate (0.05%), triethanolamine (2 ml), orthophosphoric acid (for adjusting pH value to 3.2) and B-acetonitrile in a ratio (A:B;35:65), with a flow rate of 0.8 ml/min using gradient elution. The retention time of TEL was approximately 22.60 min. The injection volume was 10 μl and column temperature was maintained at 30 ± 5°C.
Shape and morphological attributes
Polarized light microscopy: The microscopy was performed using the polarized light microscope (DM LMP-1188500, Leica Microsystems, Mumbai, India) equipped with Leica Qwin Plus software system. Small amount of sample was mounted on a glass slide and evaluated for the bi-fringence for the TEL, spray dried powder and melt extrudates.
Scanning electron microscopy: The shape and surface morphological attribute of solid dispersions were carried out using a scanning electron microscope (Philips XL-30, Netherlands). Samples were mounted on carbon sticky tabs and sputter coated with thin goldpalladium conductive layer using Polaron range sputter coater.
Physical state characterisation of solid dispersion
Differential scanning calorimetry (DSC) studies: The thermal characteristics of TEL, polymer and solid dispersions (spray dried and melt extrudates) were investigated using a differential scanning calorimeter (Modulated DSC -1 from Mettler Toledo). Samples of were placed in sealed aluminium pans. The heating temperature was in the range of 25°C to 300°C with a heating rate of 10°C/min. The results were analysed using STARe evaluation software.
Powdered X-ray diffraction (PXRD) studies: The physical state of TEL, polymer and solid dispersions (sprays dried and melt extrudates) was assessed using powder X-ray diffraction (D-8 Advance type, Bruker Instruments, Germany). The samples were exposed to Cu Kα radiation under 40 kV and 40 mA over the 2θ range from 3° to 40° at increments of 0.25º/min. The obtained diffractrogram were finally interpreted.
In vitro drug release
TEL (pure) and solid dispersions (equivalent to 40 mg) were accurately weighed and filled in a size ‘0’ hard gelatin capsule. The suitable sinkers were used while performing the dissolution testing.
Release studies performed using USP type II dissolution test apparatus (Electrolab 5 dissolution tester, Mumbai, India) in 900 ml dissolution media (water, hydrochloric acid buffer (pH 1.2), acetate buffer (pH 4.5) and phosphate buffer (pH 7.4), with a paddle speed of 75 rpm, and temperature of the media was maintained at 37 ± 0.5°C.
The sampling was performed at regular intervals (5, 10, 15, 30, 45, 60 and 90 min). The aliquots (5 ml) of media were withdrawn and volume was replenished with the same amount of fresh dissolution fluid each time to maintain the sink condition.
The samples were filtered through 0.45 μm membrane filters prior to analysis. The amount of drug was determined using UV-visible spectrophotometer (UV-1700, Shimadzu, Pharmaspec, Japan) at 296 nm.
Stability studies: The stability studies were conducted in accordance to ICH guidelines (Q1A). The melt extrudates, spray dried powder and TEL were packed in aluminium pouches and sealed. The packed samples were exposed to 100°C for 3 days, 25°C/60% RH and 40°C/75% RH for 15 days. The control samples were stored at 2-8°C in closed vials. The samples were analyzed for the drug content using developed and validated HPLC method.
Pharmacokinetic studies
Male Sprague-Dawley rats with average weight of 250 ± 20 g were fasted for 12 h prior to the experiments. Animals were housed under standard (25 ± 2°C, 60–70% humidity) laboratory conditions on normal dark and light cycle. Standard rat chow pellets and water was allowed ad libitum.
Animals were acclimatized to laboratory conditions before the test. The rats were divided into three groups (n=3). Group I was administered the drug powder (formulation A1), Group II; solid dispersions viz. formulation S1 and group III; formulation H1, in the form of suspension at a dose of 10 mg/kg per orally. Blood (400 μl) was collected from retro-orbital plexus under isoflurane anesthesia and were collected in tubes containing anticoagulant.
The blood samples were collected at an interval of 0.5, 1, 2, 4, 6, 8, 24, 48 and 72 h following oral administration. The plasma was separated by centrifugation at 5,000 × g for 10 min, at 4°C. The experimental protocols were approved by the Institutional Animal Ethical Committee (IAEC) and conducted according to the guidelines of ‘‘Committee for the Purpose of Control and Supervision of Experiments on Animals’’ (CPCSEA).
Plasma sample analysis: The internal standard (IS) albendazole (10 μL of 10 ng/ml) was added to an aliquot (100 μL) of plasma sample and vortexed for 15 seconds. To this, ethyl acetate (1.5 ml) was added and the mixture was vortexed for 5 min, followed by centrifugation for 8 min at 10,000 × g at 5°C. The organic layer (1.3 ml) was separated and evaporated to dryness at 40°C using a gentle stream of nitrogen. The residue was reconstituted in 300 μL of the mobile phase. Then the mobile phase was injected (injection volume 5 μL) into the LC-MS/MS system (AB Sciex instrument; API 4000 attached with Shimadzu UFLC (ultra-fast liquid chromatography) for analysis, equipped with Zorbax 6 SB-CN column (3.5 μm, 3.00 × 150 mm). The mobile phase consisted of acetonitrile and ammonium acetate buffer (pH 4.5) in a ratio of 70:30. The flow rate was 0.5 ml/min.
Statistical data analysis: The area under the drug concentration-time curve from zero to infinity (AUC), the elimination rate constant and half-life were calculated using a non-compartmental analysis (Win- Nonlin: professional edition, version 6.2.1) The maximum plasma concentration of drug (Cmax) and the time taken to reach the maximum plasma concentration (Tmax) were obtained directly from the plasma data. Levels of statistical significance (p<0.05) were assessed using the ANOVA test. All data are expressed as mean +/- standard deviation (SD). The obtained data was analysed using Graph pad prism.
Results And Discussion
Preparation of Solid dispersion
Spray drying is an efficient technology for solid dispersion preparation as it permits fast solvent evaporation leading to transformation of drug-polymer solution to solid drug-polymer particles [15]. The properties of resulting solid dispersion was a function of ratio of drug : polymer (as it led to formation of the best film), composition of solvent and other process variables, which include the aspirator rate, nozzle diameter, drying air inlet temperature, out let temperature and pressure inside the spraying chamber.
Preparation of solid dispersion using hot melt extrusion, involves mixing of polymer, drug and excipient blends thoroughly in the molten state, thus negating the use of any solvents for granulation. The molten polymer serves as the thermal binder [16]. Hot melt extrusion involves forcing a crystalline drug material or drug mixture with polymer blend through a die or orifice under set conditions such as temperature, pressure, rate of mixing and feed-rate, for the purpose of producing a solid solution/amorphous system.
Based on formulation and process variables of spray drying and hot melt extrusion optimization studies were performed (unpublished work). Melt extrusion and spray drying resulted in an amorphous state of the drug as confirmed using PXRD and DSC studies. The temperature of the process was set as 200 ± 5°C and 240 ± 5°C while other parameters have been kept same as mentioned in Table 1. A glassy solid solution (Figure 2) was obtained at temperature of 240 ± 5°C.
| Method | Parameters | Set |
|---|---|---|
| Hot Melt Extrusion | Screw speed | 100 rpm |
| Torque | 0.1 to 0.15 | |
| Temperature | 240 ± 5°C | |
| Spray Drying | Pressure | 30-150 mbar |
| Oxygen flow | 4 | |
| Spray | 100% | |
| Inlet temperature | 40 ± 5°C | |
| Outlet temperature | 35 ± 5°C | |
| Head temperature | 38 ± 5°C |
Table 1: Process parameters for preparation of solid dispersions.

Figure 2: Physical appearance of solid dispersions after processing.
The assay (performed using HPLC) of the drug in the solid dispersion prepared by hot melt extrusion and spray drying method was found to be 100.0% and 99.3%, respectively. Processing Soluplus® at temperature exceeding 200 ± 5°C did not show any decomposition. The retention time of Soluplus® inside the screw could be optimized to prevent degradation of soluplus® and yet facilitating formation of glassy solid solution for high melting drug such as TEL.
Solubility studies
Polyvinyl caprolactam–polyvinyl acetate–polyethylene glycol graft copolymer (Soluplus®) is a new polymer with amphiphilic properties, designed and developed for solid solutions. Soluplus® possesses bifunctional character (as a matrix polymer for solid solutions and an active solubilizer through micelle formation in water) and can be considered as a member of the fourth generation of solid dispersions [17,18]. As it is hydrophilic and non-ionic (clearly depicted from the hydroxyl groups in its structure), its solubility does not change along with the gastrointestinal tract. It is slightly surface active, a property which can be useful to maintain supersaturation of poorly soluble drugs in the gastrointestinal tract. The effect of Soluplus® in improving the aqueous solubility of TEL (0.6 μg/ml), is depicted in Table 2. The aqueous solubility of TEL was 44 fold and 20 fold higher with hot melt extrusion and spray dried solid dispersions as compared with pure drug. Formulating solid dispersions resulted in improved solubilisation of TEL at pH 1.2, pH 4.5 and pH 7.4 buffer as compared to pure drug (Table 2). However, in all instances, HME appeared more promising than spray drying in improving solubility of TEL. Latter is attributed to the solubilising capacity of Soluplus® in buffers (of pH 1.2, 4.5 and 7.4) to result in smaller particle size at a molecular level, thus providing an increased surface area. Further, the transformation of crystalline TEL into a higher energetic amorphous form [19] further enhances its aqueous solubility.
| Media Used | Solubility (mg/mL); Mean ± SD | ||
|---|---|---|---|
| Telmisartan API | Spray Dried SD | HME SD | |
| Water | 0.0017 ± 0.0001 | 0.0344 ± 0.0014 | 0.0744 ± 0.0061 |
| HCl Buffer (pH 1.2) |
0.9000 ± 0.0086 | 1.1121 ± 0.0072 | 2.9405 ± 0.0312 |
| Acetate Buffer (pH 4.5) |
0.0098 ± 0.0000 | 0.0948 ± 0.0001 | 0.1086 ± 0.0098 |
| Phosphate Buffer (pH 7.4) | 0.0020 ± 0.0001 | 0.1382 ± 0.0075 | 0.1979 ± 0.0098 |
Table 2: Solubility profile of Telmisartan and formulations (n=3) in different medias.
Shape and morphological attributes
Scanning electron microscopy (SEM) images and polarized light microscopy (PLM) of pure TEL and solid dispersions are shown in Figures 3 and 4. TEL (Figures 3a and 4a) appeared as needle crystals with smooth surfaces of homogeneous morphology as revealed from SEM and PLM micrograph. However, the solid dispersion prepared by the spray drying method showed a spherical or semispherical shape with relative loss of definite space group or symmetry, thus confirming the loss of its crystalline structure and formation of an amorphous system. Solid dispersion prepared by melt extrusion method (Figures 3c and 4c) was also amorphous and showed a planar structure. Latter is predicted to provide an increased surface area and subsequently enhances the solubilisation of the drug.

Figure 3: Depicts SEM images of a) Pure TEL and b) SD prepared by spray drying c) SD prepared by HME.

Figure 4: Polarized light microscopy images of a) TEL and solid dispersions b) spray dried and c) HME.
In vitro drug release studies
The dissolution studies of different solid dispersion prepared with Soluplus® as a polymer were performed in all pH condition and in aqueous medium. The in vitro release profile of TEL pure and the solid dispersions (spray dried and melt extrusion) in hydrochloric acid (pH 1.2) and water can be depicted in the Figures 5a and 5d.
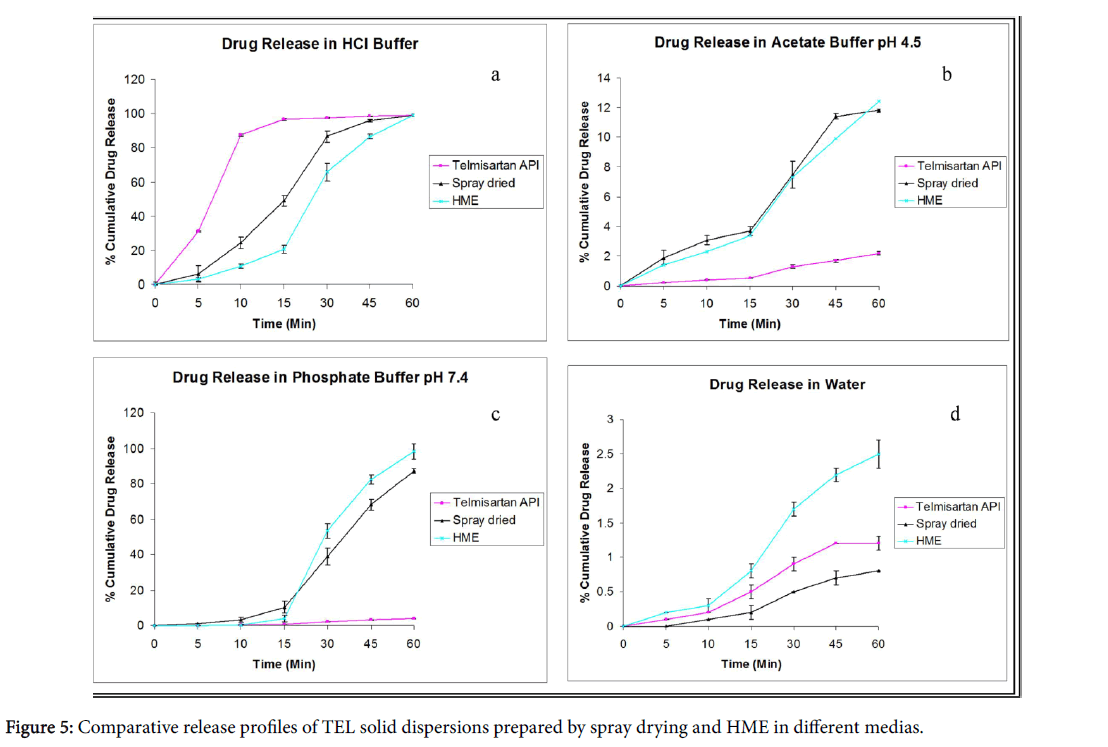
Figure 5: Comparative release profiles of TEL solid dispersions prepared by spray drying and HME in different medias.
The % drug release in water was very low (2.5% in 60 min) as compared to 100% drug release during the same time in 0.1 N HCl. The solid dispersions showed an improved dissolution (with drug release of approximately 13%) in acetate buffer (pH 4.5) and phosphate buffer (pH 7.4) as compared to pure drug (Figures 5b and 5c).
The drug release in phosphate buffer (pH 7.4) was lower in case of spray dried dispersion as compared to hot melt solid dispersion which exhibited complete and consistent release of drug.
The pure TEL did not show any significant release in phosphate buffer (pH 7.4). The results of drug release studies were in agreement with inherent properties of drug molecule under investigation [20,21]. In all cases, solid dispersions exhibited faster and almost complete dissolution compared to the pure drug and its corresponding physical mixtures.
Latter is attributed to the reduction of crystal size, a solubilization effect of the Soluplus®, absence of aggregation of drug crystallites, improved wettability and dispersibility of a drug from the dispersion, dissolution of the drug in the hydrophilic carrier, and conversion of the drug to an amorphous state [22].
Physical state characterization
Thermal characterization provides information on melting point, recrystallization, and decomposition of the polymeric system. The DSC spectra of pure drug TEL, Soluplus® and physical mixture of the drug and polymer are shown in Figure 6.
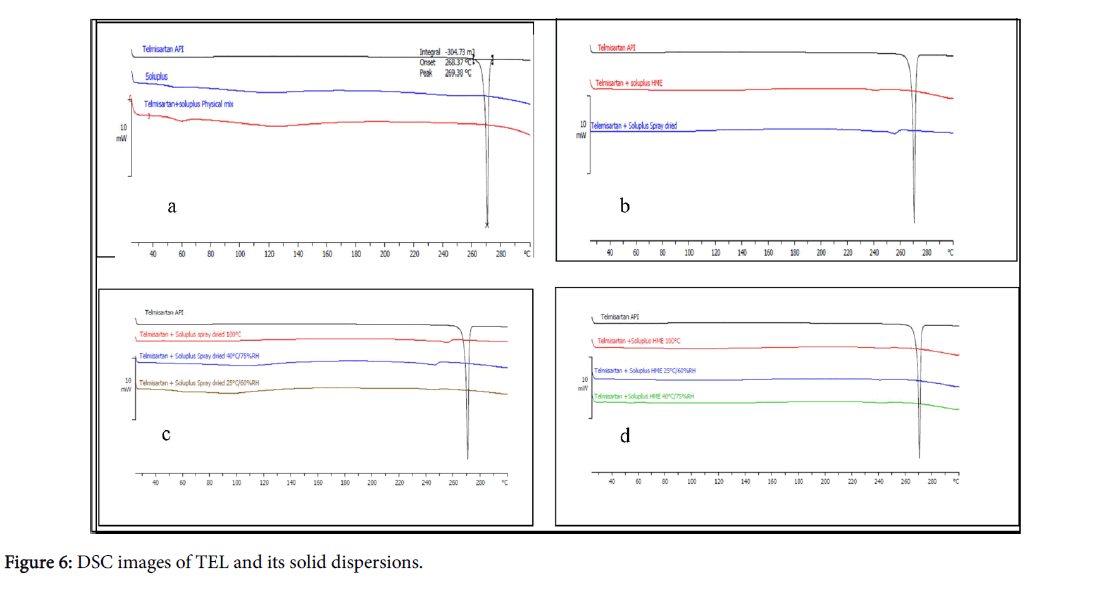
Figure 6: DSC images of TEL and its solid dispersions.
TEL had characteristic endothermic peak at 268°C which corresponds to its melting point, whereas soluplus® being an amorphous polymer did not show any sharp melting but exhibited a glass transition at about 70°C.
The absence of sharp endothermic peak in thermogram of physical mixture indicates interaction between TEL and Soluplus® which is corroborated from results of PXRD studies.
No endothermic or exothermic peaks was observed during entire temperature range from 40 to 280°C in thermograms of spray dried and hot melt extrusion solid dispersions.
The absence of drug peak in thermogram demonstrated a strong interaction between the novel grafted polymer soluplus® and drug which suggested the presence of TEL in amorphous form in the solid dispersions prepared by both methods.
Powder X-ray diffractometry (PXRD) pattern of TEL showed prominent peaks indicating the crystalline nature of the drug. The diffractograms of solid dispersions prepared by spray dried or melt extrusion revealed the amorphous nature of the prepared formulations obtained in PXRD studies, confirming stability of solid dispersions (Figure 7).
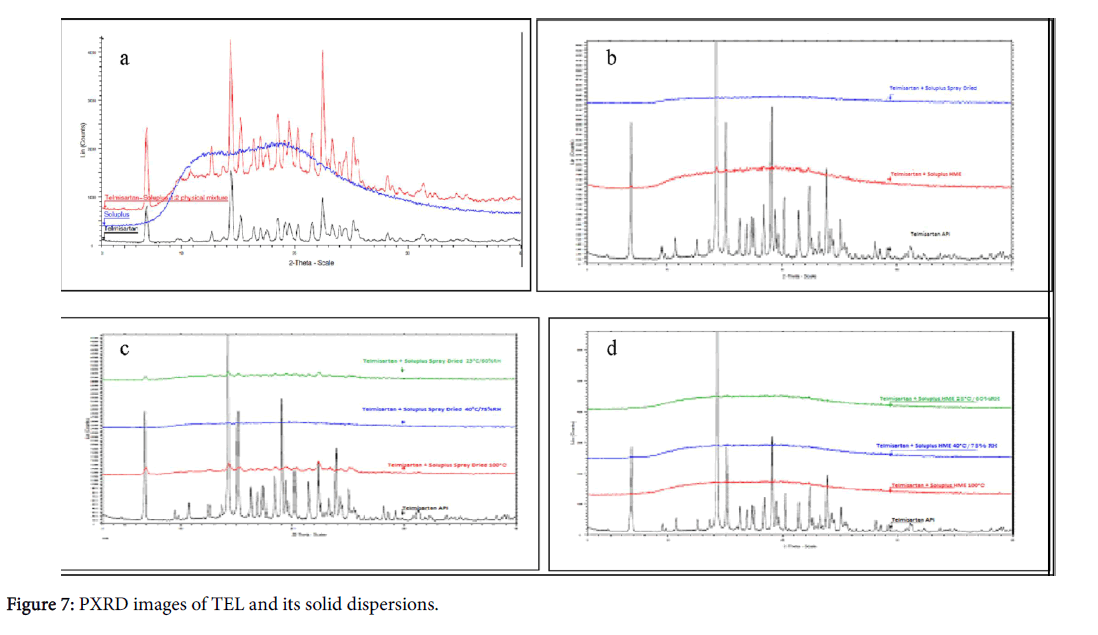
Figure 7: PXRD images of TEL and its solid dispersions.
Pharmacokinetic studies
The pharmacokinetic parameters of TEL were determined after oral administration of the pure TEL drug (10 mg/kg) and its solid dispersions (10 mg/kg), prepared by either method (spray drying and hot melt homogenization) comprising of TEL and soluplus® (ratio=1:3). The plasma concentration time profile for TEL and solid dispersions is presented in (Table 3).
| Parameters | Telmisartan Suspension | Melt extrusion dispersion | Spray dried dispersion |
|---|---|---|---|
| Cmax (µg/mL) | 419 ± 38 | 7374 ± 173 | 5555 ± 267 |
| Tmax (h) | 5.50 ± 0.5 | 0.63 ± 0.05 | 0.50 ± 0.03 |
| AUC last (h µg/mL) | 6076 ± 72 | 26468 ± 240 | 32489 ± 440 |
| t1/2 (h) | 6.92 ± 0.52 | 8.79 ± 0.64 | 13.3 ± 1.89 |
| AUC inf (h µg/mL) | 6233 ± 78 | 26601 ± 256 | 32859 ± 459 |
Table 3: Pharmacokinetic parameters of Telmisartan and solid dispersions (Mean ± SD) n=3.
The total plasma concentrations of the drug from the solid dispersion were higher than the TEL suspension. In particular up to 3h, the solid dispersion (by both the process) gave significantly higher initial plasma concentrations than the pure drug. The solid dispersions yielded significantly higher AUC, Tmax and Cmax of the drug than the TEL suspension.
The solid dispersion prepared by spray died technique showed higher AUC but the melt extrusion showed higher Cmax. However, AUC values for solid dispersions were about 4 to 5.5 fold higher than the pure drug suspension. These values clearly indicate the improvement in bioavailability of TEL from SDs when compared to its suspension, confirming an increase in solubility and dissolution rate thus enhancing the oral bioavailability.
Conclusion
The potential of Soluplus® as a polymer for enhancing the solubility of a BCS Class II drug, Telmisartan, is the highlight of the present study. Solid dispersions of TEL were amorphous in nature and possessed high solubility under varying pH range (1.2-7.4). They were stable and showed significantly higher drug release as compared to the free drug. More to the above, the methods for preparation namely, spray drying and hot melt extrusion are easily scalable and commercially feasible methods. Further, the results of the pharmacokinetic study clearly indicated the potential of solid dispersions as drug delivery systems. Thus, our intent of incorporating a poorly soluble drug into a solid dispersion for attaining enhanced bioavailability was successfully achieved.
References
- David SE, Timmins P, Conway BR (2012) Impact of the counterion on the solubility and physicochemical properties of salts of carboxylic acid drugs.Drug DevInd Pharm 38: 93-103.
- Yoshida T, Kurimoto I, Yoshihara K, Umejima H, Ito N, et al. (2012) Aminoalkyl methacrylate copolymers for improving the solubility of tacrolimus. I: Evaluation of solid dispersion formulations. Int J Pharm 428: 18–24.
- Zhang Y, Wang J, Bai X, Jiang T, Zhang Q, et al. (2012) Mesoporous silica nanoparticles for increasing the oral bioavailability and permeation of poorly water soluble drugs.Mol Pharm 9: 505-513.
- Sheth P, Sandhu H, Singhal D, Malick W, Shah N, et al. (2012) Nanoparticles in the pharmaceutical industry and the use of supercritical fluid technologies for nanoparticle production.Curr Drug Deliv 9: 269-284.
- Perrett S, Venkatesh G (2006) Enhancing the bioavailability of insoluble drug compounds, innovations in pharmaceutical technology. Drug Delivery 19: 80-85.
- Forster A, Hempenstall J, Rades T (2001) Characterization of glass solutions of poorly water-soluble drugs produced by melt extrusion with hydrophilic amorphous polymers. J Pharm Pharmacol 53: 303-315.
- Siew A (2013) Advancing polymers for solubility enhancement. Pharmaceutical Technology s6-s11
- Williams M, Tian Y, Jones DS, Andrews GP (2010) Hot melt extrusion technology: optimizing drug delivery. European Industrial Pharmacy 7: 7-10.
- Dhirendra K, Lewis S, Udupa N, Atin K (2009) Solid dispersions: a review.Pak J Pharm Sci 22: 234-246.
- Marasini N, Tran TH, Poudel BK, Cho HJ, Choi YK, et al. (2013) Fabrication and evaluation of pH-modulated solid dispersion for telmisartan by spray-drying technique.Int J Pharm 441: 424-432.
- Goldberg AH, Gibaldi M, Kanig JL, Mayersohn M (1966) Increasing dissolution rates and gastrointestinal absorption of drugs via solid solutions and eutectic mixtures. IV. Chloramphenicol--urea system.J Pharm Sci 55: 581-583.
- Sekiguchi K, Obi N, Ueda Y (1964) Studies on absorption of eutectic mixture. Ii. Absorption of fused conglomerates of chloramphenicol and urea in rabbits.Chem Pharm Bull (Tokyo) 12: 134-144.
- Chokshi RJ, Zia H, Sandhu HK, Shah NH, Malick WA (2007) Improving the dissolution rate of poorly water soluble drug by solid dispersion and solid solution: pros and cons.Drug Deliv 14: 33-45.
- Saffoon N, Uddin R, Huda NH, Sutradhar BK (2011) Enhancement of oral bioavailability and solid dispersion: A review. Journal of Applied Pharmaceutical Science 1: 13-20.
- Paudel A, Worku ZA, Meeus J, Guns S, Van den Mooter G (2013) Manufacturing of solid dispersions of poorly water soluble drugs by spray drying: formulation and process considerations.Int J Pharm 453: 253-284.
- Liu J, Cao F, Zhang C, Ping Q (2013) Use of polymer combinations in the preparation of solid dispersions of a thermally unstable drug by hot-melt extrusion, ActaPharmaceuticaSinica B 3: 263-272
- Lim H, Howland H, Fahmy R, Hoag S (2010) Solubility enhancement of poorly water soluble drug using a novel polymeric SolubilizerSoluplus®. AAPS Annual Meeting and Exposition. New Orleans, LA, PSWC.
- Leuner C, Dressman J (2000) Improving drug solubility for oral delivery using solid dispersions.Eur J Pharm Biopharm 50: 47-60.
- Al-Hamidi H, Edwards AA, Mohammad MA, Nokhodchi A (2010) To enhance dissolution rate of poorly water-soluble drugs: glucosamine hydrochloride as a potential carrier in solid dispersion formulations. Colloids Surf B: Biointerfaces 76: 170-178.
- Nakatani M, Takeshi S, Ohki T, Toyoshima K (2004) Solid Telmisartan pharmaceutical formulations. US Patent 2004/0110813 A1.
- Velasquez MT (1996) Angiotensin II receptor blockers. A new class of antihypertensive drugs.Arch Fam Med 5: 351-356.
- Craig DQ (2002) The mechanisms of drug release from solid dispersions in water-soluble polymers.Int J Pharm 231: 131-144.
Citation: Enose AA, Dasan P, Sivaramakrishanan H, Kakkar V (2016) Formulation, Characterization and Pharmacokinetic Evaluation of Telmisartan Solid Dispersions. J Mol Pharm Org Process Res 4: 131. DOI: 10.4172/2329-9053.1000131
Copyright: ©2016 Enose AA, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 17019
- [From(publication date): 6-2016 - Aug 30, 2025]
- Breakdown by view type
- HTML page views: 15726
- PDF downloads: 1293