Research Article Open Access
Production, Partial Purification and Characterization of an Extracellular Psychrotrophic Lipase from Pseudomonas Sp. ADT3
| Arpita Dey1,4*, Amarnath Chattopadhyay2, Subhra Kanti Mukhopadhyay2, Pradipta Saha2, Sabyasachi Chatterjee1, Tushar Kanti Maiti3 and Pranab Roy4 | |
| 1Department of Biotechnology, The University of Burdwan, India | |
| 2Department of Microbiology, The University of Burdwan, India | |
| 3Department of Botany, The University of Burdwan, India | |
| 4Department of Biotechnology, Haldia Institute of Technology, India | |
| Corresponding Author : | Arpita Dey Department of Biotechnology The University of Burdwan, India Tel: 0342 263 4975 E-mail: arpitadey086@gmail.com |
| Received July 22, 2014; Accepted August 28, 2014; Published August 30, 2014 | |
| Citation: Dey A, Chattopadhyay A, Mukhopadhyay SK, Saha P, Chatterjee S, et al. (2014) Production, Partial Purification and Characterization of an Extracellular Psychrotrophic Lipase from Pseudomonas Sp. ADT3. J Bioremed Biodeg 5:242. doi:10.4172/2155-6199.1000242 | |
| Copyright: © 2014 Dey A, et al. This is an open-a ccess article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. | |
| Related article at |
|
Visit for more related articles at Journal of Bioremediation & Biodegradation
Abstract
Psychrotrophic Pseudomonas ADT3 (NCBI GenBank Acc.no.JX914667) is capable of growth on lipid as the sole carbon source. In this paper, we report the purification and characterization of an extracellular lipase from psychrotroph, isolated from soil sample of Ny- Alesund, Svalbard, Arctic region. The Pseudomonas ADT3 isolate produces lipase enzyme in the extracellular minimal media with only 1% olive oil. The lipase was purified from the concentrated culture supernatant. The crude enzyme was partially purified by saturated ammonium sulphate precipitation followed by extensive dialysis. Enzyme activity was found to be induced 6-folds in presence of 1.2 mM lead ion but strongly inhibited by heavy metals Hg2+ as well as EDTA and β-mercaptoethanol. The purified lipase has activity at two pH optima of pH i.e. pH 3.5 and 8.5. Optimum temperature for lipase activity was recorded at 22cC. The purified active fraction of lipase exhibits specific activity of 527.8 U/mg. The Vmax and Km was 144.93 U/mg/min and 0.260 mM respectively determined using Lineweaver-Burk plot. Zymogram analysis revealed prominent lipase band at 13.9 kDa range in the 80% saturated ammonium sulphate purified enzyme fraction.
| Keywords |
| Psychrotroph; Arctic; Extracellular lipase; Zymogram |
| Introduction |
| Lipases (triacylglycerol acylhydrolase, EC 3.1.1.3) constitute one of the most important groups of biocatalysts for biotechnological applications. They can catalyze both the hydrolysis and the synthesis of esters from glycerol and long-chain fatty acids [1]. Recently, coldactive lipases from different microorganisms have been found to be useful for industrial application, further discovery of new lipases with commercially useful properties still attracts a great deal of interest [2,3]. They have a wide range of potential applications in the hydrolysis, esterification, and trans-esterification of triglycerides and in the chiral selective synthesis of esters [4,5]. These enzymes have high catalytic efficiencies at low temperature, unusually high values for Kcat and for catalytic efficiency (kcat/Km), and lower thermal stability, which are expected to apply in many different industrial fields, cleaning detergents, leather processing, food industry, and biotransformation at low temperatures [2]. |
| Arctic region is considered the unique, mostly pristine and extreme environment with predominantly low temperatures. Microorganisms living here have a high degree of biochemical and physiological adaptation to cold and changeable climatic conditions, so they have a high potential for biotechnological applications [6]. It would be the new and potential sources of cold-active enzymes from Arctic microorganisms. |
| Bacterial lipolytic enzymes can be classified into eight families (I to VIII), and lipases in family I can be classified into six subfamilies, subfamilies I.1 to I.6 [7]. Most of the Pseudomonas lipases are classified in Subfamilies I.1 to I.3. Lipases from Subfamily I.1, with the smallest molecular mass (approximately 30 kDa), include those from Pseudomonas aeruginosa [8] and Pseudomonas fragi [9]. Subfamily I.2 lipases, which show about 60% amino acid identity to those of Subfamily I.1, consist of about 320 amino acids and have molecular masses of approximately 33 kDa. |
| Cold-active lipases have been reported in many kinds of psychrophilic or psychrotrophic microorganisms which includes Acinetobacter [10-12], Moraxella [13], Psychrobacter [14-16], Aeromonas [17], Pseudomonas [18-21], Pseudoalteromonas [22,23] and Candida [24]. Cold adapted lipase from psychrotrophic Pseudomonas sp. have also been reported, such as lipase (PFL) produced by Pseudomonas [25]; cold adapted lipase (KB-Lip) produced by a psychrotrophic Pseudomonas sp. strain KB700A [20]; cold adaptive lipase (LipP) excreted by Pseudomonas sp. strain B11-1 [18]. However, lipases from different species or even different strains of the same species may differ in their catalytic properties such as the hydrolytic and synthetic activity, pH and thermal stability, activator requirement and so forth. Thus, the search for some new lipases remains important due to the wide range of practical industrial applications, which requires the availability of lipolytic enzymes displaying unique and specifically desired properties. |
| In this communication, we report the biochemical characterization and purification of an extracellular cold active novel lipase from Pseudomonas sp.ADT3 isolated from soil samples collected from Ny- Alesund (78°55’N, 11°54’E) Svalbard, Arctic region during the Indian Arctic Expedition 2009, organized by National Centre for Antarctic and Ocean Research, Goa. This novel lipase is induced by lead from Pseudomonas sp. ADT3 isolate that utilizes olive oil as the sole carbon source. A simple titrimetric method was used to measure enzyme activity. Optimisation of the protocol for activity staining is also reported. The present communication would make this enzyme useful with potential applications especially in low temperature bioremediation and food processing. |
| Materials and Methods |
| Chemicals |
| All chemicals used were of analytical grade, obtained from Himedia (India) and Sigma-Aldrich Company. |
| Sample collection |
| The soil samples were collected from Ny- Alesund (78°55’N, 11°54’E) Svalbard, Arctic region during the Indian Arctic Expedition 2009, organized by National Centre for Antarctic and Ocean Research, Goa under the Ministry of Earth Science, Govt of India. |
| Isolation of lipolytic bacteria |
| Pseudomonas sp. ADT3 was isolated in our laboratory from the soil samples collected from Arctic. Soil samples have been serially diluted and plated on M9 minimal media containing 0.3%(w/v) KH2PO4, 0.6%(w/v) Na2HPO4, 0.05% (w/v) NaCl, 0.1% (w/v) NH4Cl, 2 mM MgSO4, 0.1 mM CaCl2, with 1.0% olive oil as the sole carbon source and 2% (w/v) agar [18] at pH 8.0 by spread plate method. Plates were incubated for 48 h at 22°C. Pure cultures of the isolate were maintained on M9 minimal medium agar slants containing olive oil and were routinely maintained by sub culturing. |
| Screening of isolates for lipase activity |
| Pseudomonas strain ADT3 was screened by qualitative plate assay. The strains were grown on olive oil agar base plate at 22°C for 48 h. Zone of clearance was observed maximal with ADT3 strain which was selected for further analysis. The zone of clearance was observed due to hydrolysis of olive oil to fatty acids [26]. Lipase production was monitored by irradiating plates containing fluorescent dye Rhodamine B with UV light at 350 nm. After 48 h of incubation bacterial colonies began to show an orange fluorescence, with continuing incubation time orange fluorescent halos were formed around the colonies. The orange fluorescence under UV illumination indicates the presence of lipolytic activity of ADT3 [27]. |
| Preparation of crude enzyme extract |
| ADT3 cells were grown in Erlenmeyer flasks (250 mL) with 50 mL of M9 minimal medium containing 1% olive oil as substrate and flasks were incubated in shaker incubator with 120 rpm agitation at 22oC for 48 h. Cells were harvested by centrifugation at 10,000 rpm for 10 minutes at 4°C. The supernatant obtained was filtered using 0.45 μm cellulose acetate filter membrane (Sigma-Aldrich). The cell free filtrate was used as the extracellular crude enzyme. The cell pellet obtained was suspended in 1.0 mL solution I (10 mM EDTA pH 8, 50 mM glucose, and 25 mM Tris-HCl, pH 8) with 100 μL 10 mg/mL lysozyme. The suspension was vortexed and kept at 4°C for 15 minutes followed by temperature shock at 37°C for 1 hour and further incubated at 4°C for 30 minutes. The suspension was centrifuged at 5000 rpm for 5 minutes at 4°C and the supernatant was stored as crude enzyme extract at −20°C for further studies as intracellular enzyme extract [28]. |
| Partial purification of extracellular lipase |
| Ammonium Sulphate Fractionation and Dialysis against Buffer (Desalting): The online program ammonium sulphate calculator from EnCor Biotechnology, Inc, Gainesville, Florida [29], was used for calculating the amount of solid ammonium sulphate to be added for getting desired fractions. Precipitation was done at 10%, 10-20% and 20-30% saturation for 2-3 h. and at 40-50%, 50-60%, 60-80%, and 80- 90% saturation of ammonium sulphate for overnight incubation, with magnetic stirrer at 4°C. After each precipitation step, the fraction was centrifuged at 12,000 rpm for 15 minutes at 4°C. All the precipitates obtained were resuspended in a minimal amount of buffer (10 mM Tris-HCl, pH 8.0). Then it is dialyzed against large volume of the same buffer by successive change in buffer after 2 hours at 4°C. The process was continued until the last trace of ammonium sulphate was removed. All the concentrated fractions were subjected to protein and enzyme activity assay to choose the fraction containing maximum activity. Activity staining was also done for confirmation with alpha- napthyl acetate as substrate for lipase. |
| Acetone precipitation: The crude enzyme was subjected to different fractions of acetone precipitation with continuous stirring with magnetic stirrer overnight at 4°C. The resulting precipitate was centrifuged (15,000xg) for 20 min at 4°C. The acetone precipitates were dissolved in minimal amount of 10 mM Tris HCl buffer of pH 8.0, and then dialyzed extensively against the same buffer overnight at 4ºC. The enzyme activities in the dialysate were again assayed. |
| Lipase assay |
| Extracellular lipase activity in bacterial culture supernatants after centrifugation (12,000 g for 20 min) was determined by titrimetric assay using olive oil as substrate with pH- stat autotitrator system. Lipase activity was examined by titrating free fatty acids liberated from triglycerides with alkali [30,31]. The reaction mixture contained 1 ml of 0.1 M Tris- HCl buffer pH 8.0, 50 mM KCL, 200 μl Tween 20, 1 ml olive oil or other oil as substrate, 1 ml of culture supernatant at 22°C for 3 h. The reaction mixture was mixed well on a reciprocal shaker at 60 rpm. After incubation, the mixture was shaken vigorously with 3 mL of ethanol to stop the enzyme reaction and break down the emulsion. The amount of fatty acids liberated during the reaction was estimated by titrating with 10 mM NaOH on an autotitrator system using phenolphthalein indicator. Blank contained the same components except enzyme solution. One unit of lipase activity was defined as the amount of enzyme required to liberate 1 μM equivalent of free fatty acid per minute. Specific activity was defined as units of lipase activity per milligram protein. Specific activity was measure by using following formula. The results obtained were means of triplicates for each experiment. |
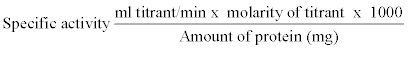 |
| Optimization of different parameters for enzymatic activity |
| The different parameters like substrate, pH, temperature, incubation time and cofactors were optimized for enzymatic activity. During optimization of each parameter, all other factors were kept unaltered. Enzyme was extracted as described above. |
| Substrate preference in reaction mixture for performing lipase activity was measured by the amount of fatty acids liberated by titrating with 10 mM NaOH using phenolphthalein indicator. Substrates used were mustard oil, coconut oil, sunflower oil, olive oil and tributyrin at 1% (v/v) concentration. |
| For the determination of pH stability the pH of the reaction mixture was varied with different buffer systems, citrate- phosphate buffer for pH 2.0 - 6.0, phosphate buffer for pH 6.5- 7.5 and Tris-HCl buffer for pH 8.0-11.0 respectively for lipase activity assay. For each pH, a blank without the enzyme was carried out and the values were subtracted from the experimental ones. |
| The optimal temperature for activity was measured from 10°C- 80°C at pH 8.5 for 3 h using the standard assay method. For the determination of thermal stability, the purified lipase was incubated for 30, 60, 90,120, 150, 180, 210, 240 min at 20°C, 30°C, 50°C and 60°C and 70°C respectively, then the residual lipase activity was measured as described in the standard assay method. |
| For determining the effect of incubation time on enzymatic activity the reaction mixture was incubated for various time interval i.e.30 min, 60 min, 90 min, 120 min, 150 min, and 180 min respectively at 22°C. |
| The effects of various compounds and inhibitors on lipase activity were examined. The different cofactors like calcium ion, magnesium ions, zinc, iron, lead, silver, EDTA etc. at 1.0 mM concentration were added. |
| Determination of kinetic parameters |
| The kinetic parameters Km and Vmax were determined from Lineweaver Burk double reciprocal plots of substrate concentration vs. initial reaction rates [32]. The Michaelis-Menten constant (Km) of purified extracellular lipase was determined by varying the concentration of olive oil. The initial velocity measured by quantitatively measuring the amount of the product at various time intervals [33]. |
| Extracellular protein profile of the isolate |
| Extraction of extracellular proteins: The cells were grown in two separate flasks, one in M9 minimal media containing 1% (v/v) olive oil and in other set M9 with 1% (w/v) peptone for 48 h with shaking at 22°C. The cells were harvested by centrifugation at 12,000 rpm for 10 min. The cell free supernatant was collected. The extracellular proteins were precipitated by 30% acetone overnight at 4°C. Finally the proteins were dissolved in 0.1 M Tris-HCl buffer pH 6.8. |
| Protein determination: The protein content of the extract was determined following the method of Bradford [34]. |
| Determination of molecular mass: Molecular mass of the Pseudomonas ADT3 strain producing lipase was determined by SDSPAGE [35] using a 12% resolving and 5% stacking gel. 50 μg of protein was loaded in each well. For SDS-PAGE, standard molecular weight markers were used. The gel was stained with Coomassie Blue R 250 and destained with 30% methanolic destaining solution. The gel was observed in the gel documentation system (Vilbur Lourmat, France) and molecular mass was analysed with Quantum Capt software. |
| Immunisation of Rabbit to Raise Specific Antibody |
| A single oil inducible band of 13.9 kDa was cut from the gel and homogenised by crushing with a sterile glass rod in Eppendrof tube containing 500 μl autoclaved water. A rabbit was injected subcutaneously at 5 to 6 different sites with this protein homogenate mixed 1:1 with Freund’s complete adjuvant twice (once per month) followed by two more injections of the same protein homogenate mixed 1:1 with Freund’s incomplete adjuvant. |
| Dot Blot: To check serum antibody titre, dot blot assay was done. 2 μl of the different dilutions (1:10, 1:100 and 1:1000) of antigen i.e., total bacterial protein obtained from oil grown cells was spotted onto PVDF membrane. 1 mg/ml BSA in different volumes (1 μl, 2 μl, 3 μl) was also spotted on the membrane as control. |
| Western Blot: The SDS PAGE gel, after electrophoresis (unstained) was electroblotted onto PVDF membrane (Sigma Aldrich USA) at 45 volts for 3 hours. The PVDF membrane was blocked with 3% milk powder for 1 hour at room temperature and washed thrice with buffer A(10 mM tris HCl pH8, 1 mM EDTA pH8, 0.05% tween 20 and 0.9% NaCl), followed by incubation in 1:100 dilution of antiserum in the same buffer, at 4°C overnight. Then the membrane was washed with buffer A thrice, five minutes each and incubated in 1:15,000 dilutions of Goat anti-rabbit IgG coupled to alkaline phosphatase (Sigma Aldrich USA) in buffer A for 2 hours at room temperature. The membrane was again washed in buffer A, 5 minutes each, for three times and equilibriated for 30 minutes in alkaline phosphatase buffer (100 mM tris HCl pH 9.5, 100 mM NaCl and 5 mM MgCl2). The membrane was stained with BCIP/ NBT (5-Bromo, 4-Chloro, 3-Indolyl phosphate/Nitrobluetetrazolium) in alkaline phosphatase buffer and kept in the dark. |
| Preadsorption of the Serum Antibody: 10 μl of the peptone grown cellular proteins were spotted on small strips of nitrocellulose membrane and air dried. These strips were immersed in 1:100 dilution of the antiserum in buffer A and incubated by gentle shaking for 1 hour. The strips were removed and the antiserum thus obtained contained antibodies specific only for oil induced proteins. This process was repeated until all the antibodies against the common antigens were removed. |
| Native PAGE for Zymogram Analysis |
| For zymogram studies, ADT3 cells were grown in M9 minimal medium with 1.0% olive oil for 48 hours. Extracellular proteins were extracted by ammonium sulfate precipitation and partially purified, and hence the protein concentration was measured by the Bradford assay. About 100 μg of proteins was resolved by 10% native gel [36] under non- denaturing condition. Electrophoresis was carried out at 4°C at 150 V of constant voltage. One part of gel was stained with coomasie blue and the unstained counterpart of the gel was assayed in situ for lipase activity by using a-naphthyl acetate (a-NA). Lipolytic active zone was identified by staining the gel with 0.03% Fast Blue RR salt, 0.05% α-naphthyl acetate, and 1% acetone in 25 mM Tris–HCl buffer at pH 7.4 [37]. |
| Statistical analysis |
| For statistical analysis, standard deviations for each of the experimental results were calculated using Microsoft Excel software. Results were expressed as the means of three independent determinations. Significance has been presented in the form of probability (p - 0.05) values |
| Results and Discussion |
| Isolation and identification of psychrotrophic lipase producing bacteria |
| Among numerous colonies detected, 10 colonies were randomly selected from the culture plate with lipid as sole carbon source. Most of the colonies were colourless with a few forming pigments. Among 10 isolates, one strain ADT3 showed maximal zone of clearance in olive oil agar base plate (Figure 1a) which is further confirmed by Rhodamine B plate assay where orange fluorescence under UV illumination indicates the presence of lipolytic activity of ADT3 (Figure 1b). |
| Optimization of conditions for enzymatic activity |
| Effect of pH on lipase activity: Two pH optima for enzymatic activity were noted at pH 3.5 (acidic) and pH 8.5 (alkaline), respectively. |
| Figure 2 clearly shows that first, there is a sharp increase in enzyme activity at pH 3.5 which drops around neutral pH followed by a peak at pH 8.5 that gradually falls into a plateau. The most probable explanation may be the presence of two isoforms of the same lipase enzyme. |
| The pH-stability profile made them useful for their application in industrial processes to be carried out at a pH range close to alkalinity. |
| Effect of temperatures on lipase activity: The specific activities of the enzyme were determined at various temperatures from 10-80°C. Optimum lipase activity was observed at 22°C as shown in Figure 3. In recent studies, some cold-active lipase showing temperature optima of 20–25°C [38].As shown in Figure 4, the thermal stability of lipase was assessed for several time periods at various temperatures (20–70°C). The stability of the lipase was very high at 20°C, enzyme activity decreased slightly when temperature was at 30°C. It retained half of its activity after 150 mins and 90 min incubation at 50°C and 60°C respectively. Lipase activity was completely inactivated at 70°C at 120 mins. This enzyme is more stable than other lipase from Pseudomonas sp. The rapid inactivation at high temperature of lipase was a typical property of cold-adapted enzymes. Lipase enzyme from Pseudomonas ADT3 was a little more stable than lipases from other psychrotrophs at high temperature; e.g., Pseudomonas sp. Strain KB700 [20], Pseudomonas fragi [25], Pseudomonas sp. Strain B11-1 [18]. Heat treatment at 60°C for 5 min resulted in a 70% decrease in the activity of KB-lip [20].These results indicated that this lipase was a typical cold-active enzyme. |
| Effect of various cofactors and inhibitors on lipase activity: 1000 μL supernatant (0.9 mg extracellular proteins) was used for characterization of enzyme activity. Figure 5 shows the effect of 1 mM of different metal cofactors on enzymatic activity plotted with respect to specific activity (U/mg). In the presence of Mg2+, Zn2+, Fe2+ metal ions, the activity was similar to that of the control. Ca2+ increased the lipolytic activity to some extent. The activity of lipase was found to be increased slightly in presence of lower concentration of Ca2+ but at significantly higher concentration of Ca2+, the activities were decreased slightly which is consistent with the results reported elsewhere [39]. |
| The primary role of Ca2+ seems to be to remove the released fatty acid as its calcium salt but the catalytic effect of calcium on a lipase derived from Humicola lanuginosa was explained by the removal of free fatty acids from the interface [40]. In a calcium free system, the lipase cannot adsorb at the water-fat interface, and consequently no lipolytic activity occurs. Possibly, the calcium ions compensate for the electrostatic repulsion created between the enzyme and the substrate. Chromium also is enhancing the lipase activity to some extent. Lead was found to enhance lipase activity by 6.0- fold in comparison to other metals. Optimum lead concentration was found to be 1.2 mM. The actual mechanism of action of lead ions in enzymatic activity is not clear. Probably lead ions bind to the core region of lipase in such a way that it leads to a change in conformation of active site. There are no reports for Pseudomonas sp. where Pb2+ play role in enhancing lipase activity. But few reports with mammalian system establish the role of Pb2+ ion in enhancing lipase activity. Lead activates Protein Kinase C in Immature Rat Brain Microvessels [41]. Pb2+stimulated phosphorylation of Electrophorus electricus electroplax (Na+ and K+)adenosine triphosphatase [42]. Tin and cadmium also increased enzyme activity to some extent i.e 3.0-fold and 2.0- fold respectively. The lipase activities were inhibited by heavy metals such as Zn2+ and Hg2+ .Hg2+, Zn2+ have been reported to have inhibitory effect on Pseudomonas lipases [43,44]. This is likely due to the proximity of the -SH group to the catalytic and interfacial binding site but partially remote from the catalytic site which may have induced the marked loss of activity [45,46]. The catalytic effect of lipases might be considered to be of Ser, His, and Glu or Asp, thus the bulky Hg2+ group might cause steric interference to the approach of the substrate to the active site. Ag3+, EDTA [20] and SDS [47] also have inhibitory effect which might be due to the removal of metal ions located on or near the active site. Olive oil as suitable substrate in enzymatic reaction was also optimized, as shown in Table 1. |
| Table 1 is listed with the effect of substrates, temperature, pH, and optimum cofactor concentrations of the enzyme taken from culture supernatant. Olive oil was found to enhance enzymatic activity than any other substrate as evident from the enzymatic rate calculations: Vmax for olive oil was 144.93 U/mg/min and Km was 0.260 mM (Figure 6). |
| Comparison of Enzyme Activity in Extracellular versus Intracellular Extracts |
| To compare the enzyme activity in the intracellular fraction obtained by lysozyme extraction of the cell pellet, the reaction was carried out under optimised conditions of 1.2 mM Pb ions, 22 °C and at pH 3.5 and pH 8.5, respectively, as shown in Figure 7. It shows enzyme activity in supernatant and cell lysate at pH 8.5 without any cofactor and with 1.2 mM Pb as cofactors. Lipase activity was found to be higher in the extracellular fraction. Percentage of total activity in supernatant fraction with 1.2 mM Pb ions after 3 hours was 87%, while activity in cell lysate with 1.2 mM Pb ions after 3 hours was 13%. Thus, percentage of total activity was maximum in the extracellular supernatant with comparatively less activity in intracellular lysate, at pH 3.5 and pH 8.5 Figure 8. |
| Purification of lipase |
| The purification procedure of the cold-active lipase produced by Pseudomonas ADT3 was summarized in Table 2. Ammonium sulphate (60-80 %) precipitation followed by dialysis resulted in increase of specific activity. |
| The purification process resulted in 2.9-fold purification and a final yield of 64.4 % of the enzyme with specific activity of 1433.8 U/mg. As shown in Figure 9, the purified lipase had a protein band with a relative molecular mass of 13.9 kDa on SDS-PAGE, as well as zymograms, indicating the enzyme activity. |
| Extracellular protein profile of the strain |
| The extracellular protein profile of lipid grown cells shows one low molecular weight of 13.9 kDa inducible band (Figure 8) which may be lipase which is faint in peptone grown cell. The band seems to be lipase which is confirmed by western blotting and activity staining. |
| Immunoblotting: As seen in the dot blot assay, the antiserum reacted with all the dilutions of oil grown proteins but no reaction was obtained with BSA (Figure 9). In the western blot analysis with the total antiserum, bands were observed in response to oil grown proteins as well as oil+ peptone grown proteins (Figure 10a and 10b). After the antiserum was preadsorbed on peptone grown proteins, response was obtained only for oil grown proteins. The preadsorbed antiserum did not react with peptone grown proteins (Figure 10c). The immunoblotting shows a dark band of 13.9 kDa on oil grown protein which hence confirms that the inducible band of 13.9 KDa shown in SDS-PAGE is of lipase. |
| Activity Staining with Partially Purified Enzyme Extract |
| Two 10% native gels were electrophoresed with the purified enzyme fractions of ammonium sulphate under same conditions. One gel was subjected to the usual staining (Coomassie blue R250) and destaining (30% methanol with 7.5% acetic acid) process (Figure 11a), while the other part of identical gels was subjected to activity staining with the purified enzyme fractions from the culture supernatant. At 20% and 40%, large amount of protein was precipitated. At 60% few proteins were salted out, while at 80%, moderate amount of protein was precipitated. Specific activity with the unpurified culture supernatant was 212.6 U/ mg after 1 hour, while 624.88U/mg activity was recorded with the 80% purified enzyme fraction (2.9-fold of purification could be achieved). One U of enzyme activity is defined as the amount of the enzyme that catalyzes the conversion of 1 micro mole of substrate per minute. |
| Figure 11b depicts the zymogram of purified enzyme fractions. A clearly visible brown activity band in the activity stain corresponding to the protein bands in the 80% purified enzyme fraction was seen on reaction with alpha napthyl acetate, a substrate of lipase. Alphanaphthyl acetate lipases accelerate the hydrolytic cleavage of alphanaphthyl acetate to form acetic acid and 1-naphthol, which couples with a diazonium salt fast blue RR salt to form a red-brown azo dye which is insoluble in water. |
| Further purification of the enzyme for subsequent crystal structure prediction needs to be done. |
| Conclusion |
| The main purpose of this study was characterization and partial purification of an extracellular cold-active lipase enzyme isolated from Pseudomonas ADT3 of Arctic soil. The enzyme was partially purified approx.2.9-fold with an overall yield of 64.4 % and specific activity of 1433.8 U/mg with 80% ammonium sulphate precipitation with extensive dialysis. The results obtained in this study show that olive oil is the most suitable substrate for maximum lipase production by Pseudomonas ADT3 strain. The enzyme exhibited maximum activity and stability at pH 3.5 and pH 8.5. The remarkable stability of Pseudomonas ADT3 lipase in this range has proved it to be a potential alkaline, a candidate for industrial applications such as detergent, leather manufacturing. Optimum lipase activity was observed at 22°C. These results indicated that this lipase was a cold-active enzyme. Lead was found to enhance lipase activity by 6.0- fold in comparison to other metals. Tin and cadmium also increased enzyme activity to some extent. Optimum lead concentration was found to be 1.2 mM. The mechanism of activation of this lipase by lead ions is not clear. But it is possible that lead ion binds to the core region of lipase in such a way that it leads to a change in conformation of active site. Lipase activity was strongly inhibited by heavy metals Hg2+as well as EDTA and β-mercaptoethanol. Comparison of enzyme activity in extracellular versus intracellular extracts shows that the percentage of total activity was maximum in the extracellular supernatant with comparatively less activity in intracellular lysate, at pH 3.5 and pH 8.5. It was a single peptide chain with a molecular weight of 13.9 kDa which was hence confirmed by western blotting and activity staining. Moreover, it had high tolerance to a wide range of NaCl concentrations (0.01–2 M).These properties were interesting compared with other cold-active lipase, which could obviously be related to the survival environment of Arctic microbe. These results indicated that this novel lipase was a new example of a cold-active lipase, which had great potential in the fields of the high salt wastewater treatment, bioremediation in fat contaminated cold environment. Further work is needed to elucidate enzyme reaction mechanism and amino acid sequence. |
| Acknowledgements |
| The authors thank the Department of Science and Technology (DST), New Delhi, Govt of India, for the grant supporting this research work. Prof P. Roy acknowledges National Centre for Antarctic & Ocean Research, Goa for facilitating the expedition to Ny-Alesund where the samples were collected. |
| Conflict of Interests |
| There is no conflict of interests between the authors or with any other person regarding any of the works reported or software used in this paper. |
References
|
Tables and Figures at a glance
| Table 1 | Table 2 |
Figures at a glance
 |
 |
 |
 |
| Figure 1 | Figure 2 | Figure 3 | Figure 4 |
 |
 |
 |
 |
| Figure 5 | Figure 6 | Figure 7 | Figure 8 |
 |
 |
 |
|
| Figure 9 | Figure 10 | Figure 11 |
Relevant Topics
- Anaerobic Biodegradation
- Biodegradable Balloons
- Biodegradable Confetti
- Biodegradable Diapers
- Biodegradable Plastics
- Biodegradable Sunscreen
- Biodegradation
- Bioremediation Bacteria
- Bioremediation Oil Spills
- Bioremediation Plants
- Bioremediation Products
- Ex Situ Bioremediation
- Heavy Metal Bioremediation
- In Situ Bioremediation
- Mycoremediation
- Non Biodegradable
- Phytoremediation
- Sewage Water Treatment
- Soil Bioremediation
- Types of Upwelling
- Waste Degredation
- Xenobiotics
Recommended Journals
Article Tools
Article Usage
- Total views: 16359
- [From(publication date):
November-2014 - Aug 31, 2025] - Breakdown by view type
- HTML page views : 11512
- PDF downloads : 4847