Targeting Prion-like Cis Phosphorylated Tau Pathology in Neurodegenerative Diseases
Received: 20-Jan-2018 / Accepted Date: 22-Jun-2018 / Published Date: 29-Jun-2018 DOI: 10.4172/2161-0460.1000443
Keywords: Traumatic brain injury; Alzheimer’s disease; Prion; Tau; Pin1
Overview of Prions and Neurodegenerative Disease
The accumulation of protein aggregates is a common feature of many neurodegenerative diseases including Alzheimer’s disease (AD), Parkinson’s disease (PD) and frontotemporal dementia (FTD) [1]. Each type of aggregate has one type of protein as its major component, with amyloid-β, hyperphosphorylated tau and α-synuclein being the most commonly observed. These proteins undergo a transformation from a soluble monomer to an insoluble, aggregated state through a number of intermediates [2]. Researchers have speculated that the protein deposits found in these neurodegenerative diseases may develop and spread throughout the brain in a manner analogous to that of aggregation of the prion protein (PrP) in transmissible spongiform encephalopathies (TSEs), such as Creutzfeldt-Jakob disease (CJD) [1,3]. Many recent studies in rodents [4-7], as well as in humans [8,9], support the notion that AD pathology propagates in a prion-like fashion [2]. However, there is no evidence suggesting that non-TSE neurodegenerative diseases, including AD, can be transferred between individuals in any case other than direct injection of diseased brain extracts, hence the use of “prionlike.”
Experiments involving intracerebral or intraperitoneal injections of AD brain extracts into susceptible mice have been shown to induce cerebral amyloidosis and associated pathology in a donor- and hostdependent manner, indicating prion-like features of amyloid-β (Aβ) [10]. The misfolded amyloid protein within diseased brain extracts appears to be able to seed the misfolding of Aβ in the new host, driving the accumulation of Aβ aggregates, a defining feature of AD, in several brain regions. The prion-like nature of Aβ pathology in humans was further verified through the autopsy of patients between the ages of 36 and 50 who had died of Creutzfeldt-Jakob disease (CJD) contracted as a result of treatment, typically in childhood, with human cadaveric pituitary-derived growth hormone contaminated with prions [11]. Several of these patients, none of whom had high-risk alleles for earlyonset Alzheimer’s, showed substantial amyloid-beta pathology, which is extremely uncommon for this age group. Because the pituitary gland shows amyloid pathology in a subset of Aβ+ patients, this discovery strongly suggests that the growth hormone was contaminated with pathogenic amyloid-beta and that the amyloid-beta was then transmitted through the same mechanism as the prions which caused the patients’ CJD [8].
For 25 years most research has focused on the amyloid hypothesis of AD pathogenesis and progression, which states that Aβ is the primary driver and neurotoxic element of AD. Recent roadblocks to the progression of Aβ-targeted therapies, along with the new concept of prion-like propagation of intracellular abnormal proteins, have brought tau into the spotlight as a potential therapeutic target [12-20]. Moreover, tau aggregation also plays a role in many other neurodegenerative disorders, collectively known as tauopathies. These include Pick’s disease (PiD), progressive supranuclear palsy (PSP), dementia with Lewy bodies (DLB), and corticobasal degeneration (CBD) [21,22]. Insight into the propagation and toxicity mechanisms of abnormally folded tau protein has the potential to offer promising new therapeutic targets for a number of disorders.
Structure And Toxicity Of Pathological Tau
Native tau has a relatively loose, unstructured protein with little α-helix and β-sheet structure. In the adult human brain, tau protein appears as six isoforms, all derived from a single gene by alternative splicing. Three of these isoforms contain three repeats (3R-τ) of a sequence thought to be involved in binding to microtubules; the other three isoforms contain an additional fourth repeat of the region, coded by exon 10 (4R-τ). The repeat region of tau is positioned between two basic, proline rich regions. Many of these prolines are preceded by a serine or threonine, allowing for phosphorylation. Dimerization due to disulfide cross-linking has been proposed to be a first step in the formation of NFTs, and only occurs when lysines in the microtubule binding repeat regions are phosphorylated [23]. This in turn disrupts tau’s function on microtubules and alters its protein stability, eventually leading to aggregation and tangle formation [24].
Neurofibrillary tangles are a neuropathological hallmark of tauopathies AD and other tauopathies and were previously believed to be the toxic species. However, recent studies have demonstrated that cell death occurs before tangle formation, meaning some earlier intermediate must be the source of tau toxicity [25]. The culmination of many years of increasing research into the toxicity of tau aggregation in neurodegenerative disease has led to the proposal that soluble, oligomeric forms of hyperphosphorylated tau (p-tau) are likely the most toxic entities in disease [21]. These p-tau oligomers are able to enter and exit cells in vitro, and are believed to be a major species responsible for propagation, although the exact mechanism is still unknown [25- 27]. Evidence suggests that these multimeric tau oligomers may act as templates for the misfolding of native tau, thereby seeding the spread of the toxic forms of the protein and initiating disease progression in a manner analogous to that of prions [28,29]. Additionally, these oligomers have repeatedly been found to induce cell death in numerous tauopathies and can propagate through the brain causing synaptic and mitochondrial dysfunction associated with memory deficits [30,31].
Physiologic tau is not capable of aggregating or causing the pathology observed in tauopathies. In order to find more effective therapeutic targets and treatments, any uncharacterized prion-like variants of tau, and other proteins associated with neurodegenerative diseases, must be identified. Our studies have led us to believe that cis p-tau is the soluble tau variant responsible for toxicity [32]. After protein phosphorylation on specific serine or threonine residues preceding a proline (pSer/Thr-Pro), the function of certain phosphorylated proteins is further regulated by a cis-trans conformational change around the Ser/Thr residue [33,34]. Cis, but not trans, pThr231-tau appears early in mild cognitive impairment (MCI) neurons and further accumulates only in degenerating neurons as AD progresses, localizing to dystrophic neurites, which are known to correlate well with memory loss [35]. Unlike trans p-tau, the cis conformation cannot promote microtubule assembly, is more resistant to dephosphorylation and degradation, and is more prone to aggregation [35]. Pin1, a peptidyl-prolyl cis-trans isomerase, binds to phosphorylated tau primarily at the Thr231 residue, catalyzing the conversion from cis to trans p-tau at that site [32-43] (Figure 1). When Pin1 becomes down regulated, the cis conformation of tau can accumulate [41].
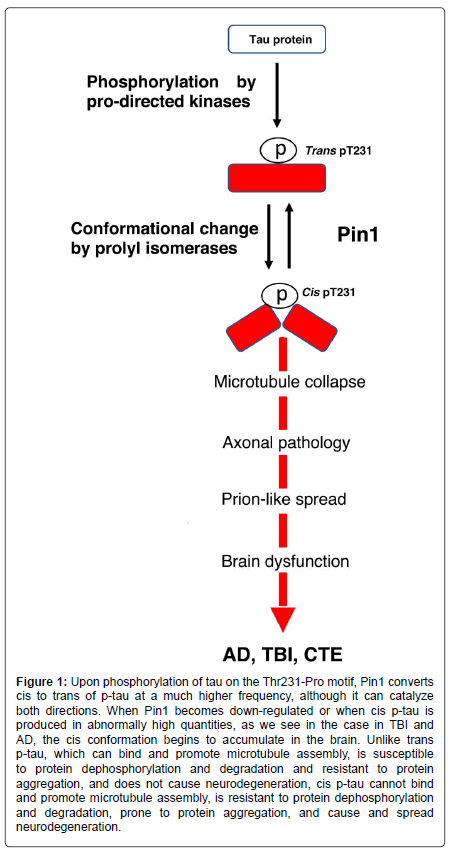
Figure 1: Upon phosphorylation of tau on the Thr231-Pro motif, Pin1 converts cis to trans of p-tau at a much higher frequency, although it can catalyze both directions. When Pin1 becomes down-regulated or when cis p-tau is produced in abnormally high quantities, as we see in the case in TBI and AD, the cis conformation begins to accumulate in the brain. Unlike trans p-tau, which can bind and promote microtubule assembly, is susceptible to protein dephosphorylation and degradation and resistant to protein aggregation, and does not cause neurodegeneration, cis p-tau cannot bind and promote microtubule assembly, is resistant to protein dephosphorylation and degradation, prone to protein aggregation, and cause and spread neurodegeneration.
Alzheimer’s Disease
The work from our lab and others has uncovered the extensive contribution of Pin1 to the development of AD pathology. Pin1 promoter polymorphisms resulting in decreased Pin1 levels are associated with an increased risk for late-onset AD [44]. In contrast, Pin1 SNPs resulting in reduced Pin1 inhibition have been associated with a delayed onset of AD [40]. In a normal human brain, Pin1 expression was relatively low in regions of the hippocampus that are susceptible to NFT-related neurodegeneration in AD (CA1 and subiculum), while Pin1 expression was higher in regions that are generally spared (CA4, CA3, CA2, presubiculum) [42]. In the brains of human AD patients, the majority of pyramidal neurons (96%) with relatively high Pin1 expression lacked tau tangles, and most pyramidal neurons (71%) with relatively low Pin1 expression had tangles [42]. Furthermore, Pin1 co-localizes and co-purifies with NFTs, and can directly restore the ability of tau to bind microtubules and promote microtubule activity [36,45]. Finally, levels of p-Thr231 tau correlate with the progression of AD, and Pin1 is strongly correlated with dephosphorylation of tau at Thr231 [43,46].
Behind all the effects of Pin1 is a conformational change between cis and trans. In 2012, by developing the first antibodies able to distinguishing the cis from trans pThr-Pro motif of tau, our group discovered that Pin1 specifically converts cis hyperphosphorylated tau, a conformation that can no longer promote microtubule assembly, to the physiologic trans [38]. Cis p-tau is more stable, less susceptible to dephosphorylation, and more prone to aggregation than trans p-tau [38]. The same study detected little to no cis or trans in healthy brains, but a dramatic increase in cis p-tau in the brains of AD patients [38]. Additionally, cis, but not trans, p-tau was strongly present in the brains of mild cognitive impairment (MCI) patients [38]. Both cis and trans p-tau were found in the cell bodies of AD neurons, but only cis p-tau was present in neuritis [38]. Besides, all cis p-tau positive cells in the hippocampus were also positive for a marker of neurofibrillary neurodegeneration, while the majority of neurons trans positive cells were negative for the marker [38]. Together these results strongly suggest that cis p-tau is an early, strong driver for AD pathology and neurodegeneration (Figure 2). In turn, the brain is protected from both cis p-tau and harmful amyloid processing by the activity of Pin1.
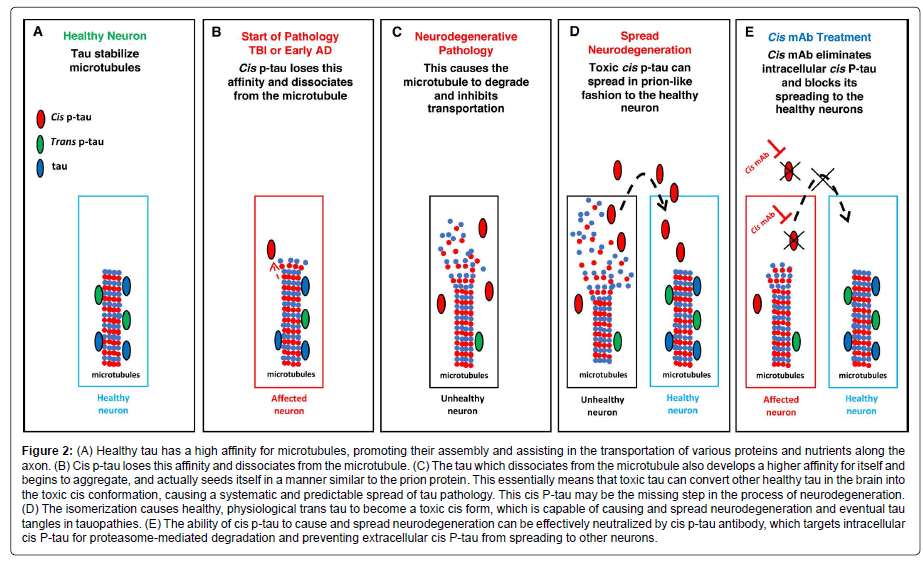
Figure 2: (A) Healthy tau has a high affinity for microtubules, promoting their assembly and assisting in the transportation of various proteins and nutrients along the axon. (B) Cis p-tau loses this affinity and dissociates from the microtubule. (C) The tau which dissociates from the microtubule also develops a higher affinity for itself and begins to aggregate, and actually seeds itself in a manner similar to the prion protein. This essentially means that toxic tau can convert other healthy tau in the brain into the toxic cis conformation, causing a systematic and predictable spread of tau pathology. This cis P-tau may be the missing step in the process of neurodegeneration. (D) The isomerization causes healthy, physiological trans tau to become a toxic cis form, which is capable of causing and spread neurodegeneration and eventual tau tangles in tauopathies. (E) The ability of cis p-tau to cause and spread neurodegeneration can be effectively neutralized by cis p-tau antibody, which targets intracellular cis P-tau for proteasome-mediated degradation and preventing extracellular cis P-tau from spreading to other neurons.
Traumatic Brain Injury and Chronic Traumatic Encephalopathy
Traumatic brain injury (TBI) and chronic traumatic encephalopathy (CTE) are closely related tauopathies that have significant potential for studying the toxicity and spread of tau in controlled conditions. Repetitive mild TBI (rmTBI), or single moderate/severe TBI (ssTBI), may cause acute and potentially long-lasting neurological dysfunction, including the development of CTE [47-49]. Additionally, TBI is an established environmental risk factor for AD [50].
The predictable and systematic nature of the progression of this tau pathology has made it possible identify 4 distinct stages of CTE. In stage I CTE, p-tau pathology is found in discrete foci in the cerebral cortex, most commonly in the superior or lateral frontal cortices, typically around small vessels at the depths of sulci. In stage II CTE, there are multiple foci of p-tau at the depths of the cerebral sulci and there is localized spread of neurofibrillary pathology from these epicenters to the superficial layers of adjacent cortex. In stage III CTE, p-tau pathology is widespread with the frontal, insular, temporal and parietal cortices showing widespread neurofibrillary degeneration. The greatest severity of this degeneration is located in the frontal and temporal lobes, concentrated at the depths of the sulci. Also in stage III CTE, the amygdala, hippocampus and entorhinal cortex show substantial neurofibrillary pathology that is found in earlier CTE stages. In stage IV CTE, there is widespread severe p-tau pathology affecting most regions of the cerebral cortex and the medial temporal lobe, sparing calcarine cortex in all but the most severe cases [51].
This systematic progression through connected brain regions is considered prion-like and quite similar to the spread of tau in Alzheimer’s. However, different specific regions of the brain are affected, and in a different order, in the two diseases. This difference could be caused by different strains of cis p-tau acting by the same mechanism, and/or by different sites of initial pathology (the entorhinal cortex in AD, and the isocortex in TBI/CTE).
By generating a monoclonal antibody (mAb) pair capable of distinguishing between cis and trans isoforms of p-tau (cis p-tau and trans p-tau, respectively), cis p-tau was identified as a precursor of tau pathology and an early driver of neurodegeneration in traumatic brain injury (TBI) and chronic traumatic encephalopathy (CTE) [52]. Notably, cis p-tau appears within hours after closed head injury and long before other known pathogenic p-tau conformations including oligomers, pre-fibrillary tangles and NFTs [52]. Neurons labeled with antibodies that recognize phospho-Thr231 and phospho-Ser262 display significantly decreased normal physiological interactions between tau and microtubules upon tau phosphorylation [53-55], which can lead to the destabilization of microtubules and eventually apoptosis (Figure 2). Importantly, other groups have independently confirmed that the pT231-tau level in human blood is a novel biomarker for acute and chronic traumatic brain injury that is correlated with poor outcome in patients [56,57].
Therapy
Studies have revealed the potential of antibody treatments in neutralizing toxic cis p-tau, thereby halting, or at least significantly delaying, neurodegeneration. This therapy has so far proved successful in mouse models of TBI [32,52,58]. Periodic treatment with a cis p-tau monoclonal antibody treatment over 4 months not only eliminated spreading of cis p-tau, axonal pathology and astrogliosis into the hippocampus without affecting physiologic trans p-tau, but also prevented tau oligomerization, tangle formation, and APP accumulation (Figure 2) [52,58]. Shorter courses of treatment (between 5 and 10 days) with delayed administration also proved effective at eliminating cis p-tau induction [58]. A humanized version of the cis p-tau antibody could prove extremely useful in treating the wide range of neurodegenerative diseases associated with toxic tau.
Multiple prion strains can exist in one patient, and the various strains often compete with each other [59,60]. This competition was recently explored as a potential therapy, where the introduction of a non- or less-toxic prion strain protected against the propagation of the toxic strain [61]. The application of this finding to the prion-like propagation of tau through overexpression of non-toxic trans-tau could be promising but requires further investigation.
Conclusion
For many years NFTs were the main subject of study in research done on tau toxicity in neurodegenerative diseases. Recently, evidence has pointed to an intermediate in the aggregation process, such as soluble cis p-tau, as a more likely candidate for the toxic species in common tauopathies. As more research has been done on tau aggregation intermediates, the prion-like nature of tau has been made apparent. Toxic tau is able to seed and spread itself through connected regions of the brain, displaying systematic and predictable patterns of spread in various tauopathies, similar to the patterns of spread seen in TSEs caused by misfolded prion protein. The difference between the tau pathology in the various tauopathies is thought to be caused by different strains or conformations of the toxic, misfolded tau. Our studies have indicated that inhibition of Pin1-mediated isomerization of cis p-tau to trans p-tau may induce tau toxicity. The use of mouse monoclonal antibodies that can distinguish trans p-tau from cis p-tau has shown promise as a potential method for both characterization and treatment of tau pathology. Once more is known about the prion-like nature of tau, and exactly how similar tau spread is to that of PrP, the strain competition model of therapy, which is currently being tested in TSEs, could potentially be applied to tauopathies. Understanding the prion-like mechanisms of the tau protein will be an important step in discovering new and more effective treatments for many common neurodegenerative diseases.
Acknowledgement
O.A. is an Alzheimer’s Association Research Fellow. The work is supported by grants from NIH (R01AG029385, R01AG046319, R01CA167677) Alzheimer’s Association (DVT-14-322623), Alzheimer’s Association, Alzheimer’s Research UK and Weston Brain Institute (MCDN-15-368711), Thome Memorial Foundation in Alzheimer’s Disease Drug Discovery Research to K.P.L, and gift donations from the Owens Family Foundation to X.Z.Z. and K.P.L.
References
- Walsh DM, Selkoe DJ (2016) A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci 17: 251-260.
- Goedert M, Masuda-Suzukake M, Falcon B (2017) Like prions: The propagation of aggregated tau and a-synuclein in neurodegeneration. Brain 140: 266-278.
- Prusiner SB (2013) Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 47: 601-623.
- Eisele YS, Fritschi SK, Hamaguchi T, Obermüller U, Füger P, et al. (2014) Multiple factors contribute to the peripheral induction of cerebral β-amyloidosis. J Neurosci 34: 10264-10273.
- Clavaguera F, Hench J, Goedert M, Tolnay M (2015) Invited review: Prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol 41: 47-58.
- Clavaguera F, Hench J, Lavenir I, Schweighauser G, Frank S, et al. (2014) Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol 127: 299-301.
- Goedert M, Falcon B, Clavaguera F, Tolnay M (2014) Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies. Curr Neurol Neurosci Rep 14: 495.
- Jaunmuktane Z, Mead S, Ellis M (2015) Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525: 247-250.
- Malkki H (2015) Alzheimer disease: Possible prion-like transmission of AD-like pathology in humans. Nat Rev Neurol 11: 612.
- Walker LC, Schelle J, Jucker M (2016) The prion-like properties of Amyloid-β assemblies: Implications for Alzheimer's disease. Cold Spring Harb Perspect Med 6.
- Brown P, Brandel JP, Sato T, Nakamura Y, MacKenzie J, et al. (2012) Iatrogenic Creutzfeldt-Jakob disease, final assessment. Emerg Infect Dis 18: 901-907.
- Hasegawa M (2016) Molecular mechanisms in the pathogenesis of Alzheimer's disease and tauopathies-prion-like seeded aggregation and phosphorylation. Biomolecules 6.
- d'Abramo C, Acker CM, Jimenez HT, Davies P (2013) Tau passive immunotherapy in mutant P301L mice: Antibody affinity versus specificity. PLoS One 8: e62402.
- Gu J, Congdon EE, Sigurdsson EM (2013) Two novel Tau antibodies targeting the 396/404 region are primarily taken up by neurons and reduce tau protein pathology. J Biol Chem 288: 33081-33095.
- Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, et al. (2013) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80: 402-414.
- Castillo-Carranza DL, Sengupta U, Guerrero-Muñoz MJ, Lasagna-Reeves CA, Gerson JE, et al. (2014) Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 34: 4260-4272.
- Sankaranarayanan S, Barten DM, Vana L, Devidze N, Yang L, et al. (2015) Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS One 10: e0125614.
- Pedersen JT, Sigurdsson EM (2015) Tau immunotherapy for Alzheimer's disease. Trends Mol Med 21: 394-402.
- Götz J, Chen F, van Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293: 1491-1495.
- Ferrari A, Hoerndli F, Baechi T, Nitsch RM, Götz J (2003) beta-Amyloid induces paired helical filament-like tau filaments in tissue culture. J Biol Chem 278: 40162-40168.
- Gerson JE, Mudher A, Kayed R (2016) Potential mechanisms and implications for the formation of tau oligomeric strains. Crit Rev Biochem Mol Biol 51: 482-496.
- Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: 239-259.
- Inoue M, Konno T, Tainaka K, Nakata E, Yoshida HO, et al. (2012) Positional effects of phosphorylation on the stability and morphology of tau-related amyloid fibrils. Biochemistry 51: 1396-1406.
- Goedert M (2015) NEURODEGENERATION. Alzheimer's and parkinson's diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 349: 1255555.
- Gerson JE, Kayed R (2013) Formation and propagation of tau oligomeric seeds. Front Neurol 4: 93.
- Goedert M, Spillantini MG (2017) Propagation of tau aggregates. Mol Brain 10: 18.
- Castillo-Carranza DL, Nilson AN, Van Skike CE, Jahrling JB, Patel K, et al. (2017) Cerebral microvascular accumulation of tau oligomers in alzheimer's disease and related tauopathies. Aging Dis 8: 257-266.
- Cowan CM, Quraishe S, Hands S, Sealey M, Mahajan S, et al. (2015) Rescue from tau-induced neuronal dysfunction produces insoluble tau oligomers. Sci Rep 5: 17191.
- Sahara N, DeTure M, Ren Y, Ebrahim AS, Kang D, et al. (2013) Characteristics of TBS-extractable hyperphosphorylated tau species: Aggregation intermediates in rTg4510 mouse brain. J Alzheimers Dis 33: 249-263.
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, et al. (2011) Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener 6: 39.
- Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, et al. (2011) Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer's disease brain. Am J Pathol 179: 2071-2082.
- Lu KP, Kondo A, Albayram O, Herbert MK, Liu H, et al. (2016) Potential of the antibody against cis-phosphorylated tau in the early diagnosis, treatment, and prevention of alzheimer disease and brain injury. JAMA Neurol 73: 1356-1362.
- Lee TH, Pastorino L, Lu KP (2011) Peptidyl-prolyl cis-trans isomerase pin1 in ageing, cancer and Alzheimer disease. Expert Rev Mol Med 13: e21.
- Lu KP, Zhou XZ (2007) The prolyl isomerase PIN1: A pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol 8: 904-916.
- Nakamura K, Zhen Zhou X, Ping Lu K (2013) Cis phosphorylated tau as the earliest detectable pathogenic conformation in Alzheimer disease, offering novel diagnostic and therapeutic strategies. Prion 7: 117-120.
- Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP, et al. (1999) The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399: 784-788.
- Albayram O, Herbert MK, Kondo A, Tsai CY, Baxley S, et al. (2016) Function and regulation of tau conformations in the development and treatment of traumatic brain injury and neurodegeneration. Cell Biosci 6: 59.
- Nakamura K, Greenwood A, Binder L, Bigio EH, Denial S, et al. (2012) Proline isomer-specific antibodies reveal the early pathogenic tau conformation in Alzheimer's disease. Cell 149: 232-244.
- Troncone L, Luciani M, Coggins M, Wilker EH, Ho CY, et al. (2016) Aß amyloid pathology affects the hearts of patients with alzheimer's disease: Mind the heart. J Am Coll Cardiol 68: 2395-2407.
- Ma SL, Tang NL, Tam CW, Lui VW, Lam LC, et al. (2012) A PIN1 polymorphism that prevents its suppression by AP4 associates with delayed onset of Alzheimer's disease. Neurobiol Aging 33: 804-813.
- Driver JA, Zhou XZ, Lu KP (2014) Regulation of protein conformation by Pin1 offers novel disease mechanisms and therapeutic approaches in Alzheimer's disease. Discov Med 17: 93-99.
- Liou YC, Sun A, Ryo A, Zhou XZ, Yu ZX, et al. (2003) Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 424: 556-561.
- Hampel H, Buerger K, Kohnken R, Teipel SJ, Zinkowski R, et al. (2001) Tracking of Alzheimer's disease progression with cerebrospinal fluid tau protein phosphorylated at threonine 231. Ann Neurol 49: 545-546.
- Segat L, Pontillo A, Annoni G, Trabattoni D, Vergani C, et al. (2007) PIN1 promoter polymorphisms are associated with Alzheimer's disease. Neurobiol Aging 28: 69-74.
- Ramakrishnan P, Dickson DW, Davies P (2003) Pin1 colocalization with phosphorylated tau in Alzheimer's disease and other tauopathies. Neurobiol Dis 14: 251-264.
- Hamdane M, Dourlen P, Bretteville A, Sambo AV, Ferreira S, et al. (2006) Pin1 allows for differential tau dephosphorylation in neuronal cells. Mol Cell Neurosci 32: 155-160.
- McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, et al. (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136: 43-64.
- Smith DH, Johnson VE, Stewart W (2013) Chronic neuropathologies of single and repetitive TBI: Substrates of dementia? Nat Rev Neurol 9: 211-221.
- DeKosky ST, Blennow K, Ikonomovic MD, Gandy S (2013) Acute and chronic traumatic encephalopathies: Pathogenesis and biomarkers. Nat Rev Neurol 9: 192-200.
- Nordström P, Michaëlsson K, Gustafson Y, Nordström A (2014) Traumatic brain injury and young onset dementia: A nationwide cohort study. Ann Neurol 75: 374-381.
- McKee AC, Stein TD, Kiernan PT, Alvarez VE (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25: 350-364.
- Kondo A, Shahpasand K, Mannix R, Qiu J, Moncaster J, et al. (2015) Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 523: 431-436.
- Augustinack JC, Schneider A, Mandelkow EM, Hyman BT (2002) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathol 103: 26-35.
- Hyman BT (2014) Tau propagation, different tau phenotypes, and prion-like properties of tau. Neuron 82: 1189-1190.
- Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E (1993) Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 11: 153-163.
- Tagge CA, Fisher AM, Minaeva OV, Gaudreau-Balderrama A, Moncaster JA, et al. (2018) Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 141: 422-458.
- Rubenstein R, Chang B, Yue JK, Chiu A, Winkler EA, et al. (2017) Comparing plasma phospho tau, total tau, and phospho tau-total tau ratio as acute and chronic traumatic brain injury biomarkers. JAMA Neurol 74: 1063-1072.
- Albayram O, Kondo A, Mannix R, Smith C, Tsai CY, et al. (2017) Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat Commun 8: 1000.
- Schoch G, Seeger H, Bogousslavsky J, Tolnay M, Janzer RC, et al. (2006) Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med 3: e14.
- Morales R, Abid K, Soto C (2007) The prion strain phenomenon: Molecular basis and unprecedented features. Biochim Biophys Acta 1772: 681-691.
- Asante EA, Smidak M, Grimsham A, Houghon R, Tomilnson A, et al. (2015) A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 522: 478-481.
Citation: Albayram O, Angeli P, Bernstein E, Baxley S, Gao Z, et al. (2018) Targeting Prion-like Cis Phosphorylated Tau Pathology in Neurodegenerative Diseases. J Alzheimers Dis Parkinsonism 8: 443. DOI: 10.4172/2161-0460.1000443
Copyright: ©2018 Albayram O, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permitsunrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 6023
- [From(publication date): 0-2018 - Sep 24, 2024]
- Breakdown by view type
- HTML page views: 5268
- PDF downloads: 755