An Approach to Computer Aided Drug Design of some Bioactive Cinnamoyl Hydrazones, In Silico and Docking Studies as Possible COX-2 Selective Inhibitors
Received: 21-Jul-2016 / Accepted Date: 02-Aug-2016 / Published Date: 09-Aug-2016 DOI: 10.4172/2155-952X.1000240
Abstract
Recently structural analogue-based drug discovery has become an important tool for designing more potent drugs. This study uses SAR, pharmacophore study and structural analogue-based novel drugs to design selective inhibitor molecules using internet-based tools. Substituted acyl hydrazones are known for wide variety of biological activities, such as analgesic, anti–inflammatory, anti-microbial, anti-convulsant, anti-platelet, anti-tubercular, antiviral, schistomiasis and anti-tumoral activities. In the last decade several cinnamic acid derivatives were reported as potent lipoxygenase inhibitors, antioxidants and anti-inflammatory agents. We found it interesting to combine these particular molecules and design cinnamoyl hydrazones, a series of N-[(1E)-2-substituted phenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl} eth-1-en-1-yl] benzamides. The designed compounds were predicted for their drug likeness and oral bioavailability. Selected compounds were docked with COX-2 enzyme and found that they were showing similar interactions as that of SC-558, 1000 time selective standard COX-2 inhibitor.
Keywords: Hydrazones, Cinnamoyl, COX-2, Docking, Oral bioavailabity, Docking studies
249394Introduction
Web-based tools offer many advantages for processing chemical information, most notably ease of use and high interactivity. The methods to calculate various molecular properties relevant to drug design are described, including the prediction of intestinal absorption, blood-brain barrier penetration, efflux and water solubility. Information about the web technology used is also provided [1].
Medicinal importance of acyl hydrazones
Acyl hydrazones are a versatile class of nitrogen-substituted molecules with a high degree of chemical reactivity, used as precursors and intermediates of many important organic molecules such as heterocyclic, pharmaceuticals, polymers, dyestuffs and photographic products [2]. Substituted acyl hydrazones show a variety of biological activity, such as analgesic, anti–inflammatory, anti-microbial, anticonvulsant, anti-platelet, anti-tubercular, anti-viral, schistomiasis and anti-tumor activities [3,4].
Medicinal importance of cinnamic acid derivatives
In recent years, intensive research has been conducted on cinnamic acid derivatives, seeking to create new polyfunctional drugs based on them [5,6]. This class of compounds is of additional interest because some representatives cinnamic acids such as caffeic, ferulic acid, etc. are widely represented in plants and together with flavonoids, participate in the regulation of many biochemical processes. The increased interest in this group of compounds is due to the variety of their pharmacological activities: e.g. antioxidative [7], antitumor [8] and antimicrobial [9], as well as to their ability to regulate the activity of a number of enzyme systems that participate in cell activation processes. Caffeic and ferulic acids inhibit 5- and 12-1ipoxygenases [10], reduce the level of leukotrienes in tissues and may potentially be used to treat bronchial asthma [11].
Selection of target
Selective inhibition of COX-2 (Cyclooxygenase-2) by NSAIDs (Non-steroidal anti-inflammatory drugs) has been proposed as an approach to reduce their associated side effects while maintaining efficacy. The improved safety profile of selective COX-2 inhibitors will allow more widespread and sustained use than is currently possible with standard NSAIDs. COX-2 is expressed after an inflammatory stimulus and releases metabolites that are used to induce pain and inflammation. Adding interest are the findings that COX-2 deletion or inhibition may lead to reduction or exacerbation of the inflammatory response, depending on the model. COX-2 appears to play a significant role in resolution of inflammation, a role that is important in the healing of gastric ulcers. During normal physiology, COX-2 levels are undetectable whereas during periods of acute and chronic inflammation, the level of COX-2 is significantly higher [12].
Based on the literature review on the anti-inflammatory and related activities of cinnamic acid derivatives [13-18] and method followed by Morphy et al. [19,20] it was aimed to design new cinnamic acid hybrids, cinnamoyl hydrazones that might act as effective bioactive agents and to evaluate the in silico parameters to optimize the activity through systematic modification of the substituent on the benzylidene core. Yet another goal of the study was to unravel the selective inhibitory activity on COX-2 enzyme by conducting docking studies for active compounds to investigate the nature of interactions contributing to the possible biological activity.
Methodology
To design new cinnamic acid-based drug candidates, the cinnamic acid part as the enoyl-acyl backbone for the hydrazones resulting in a series of N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl} eth-1-en-1-yl] benzamides [1-14]. Chemical structures were drawn and saved as Mol and Mol 2 files using Marvin sketch version 5.12.1 downloaded from Chemaxon website and chemical names (SMILES, traditional and IUPAC names) were identified [21].
Computation of molecular descriptors such as partition coefficient, topological surface area, number of hydrogen bond acceptors and donors, was carried out using Mol inspiration online tool [22]. Mol inspiration offers a broad range of cheminformatics software tools supporting molecule manipulation and processing, including SMILES and SDfile conversion, normalization of molecules, generation of tautomers, molecule fragmentation, calculation of various molecular properties needed in QSAR, molecular modelling and drug design, high quality molecule depiction, molecular database tools supporting substructure and similarity searches. Mol inspiration tools are written in Java, therefore can be used practically on any computer platform.
Using these parameters the compounds were checked for their compliance with Lipinski rule of five [23]. Drug-likeness score were computed using Mol soft website [24]. Mol soft offers software tools and services in lead discovery, modelling, cheminformatics, bioinformatics, and corporate data management; and forms partnerships with biotechnology and pharmaceutical companies. The drug-likeness scores were analyzed by comparing with the earlier reports [25]. Bioactivity score predictions were also done using Mol inspiration online tool.
Pharmmapper serves as a valuable tool for identifying targets for a novel synthetic compound, a newly isolated natural product, a compound with known biological activity or an existing drug whose mechanism action is unknown [26]. Suitable targets were predicted by Pharmmapper and docking studies were conducted using Swiss Dock software [27].
Results and Discussion
Design and nomenclature
Prompted by the medicinal importance of hydrazones and cinnamoyl moieties, the structural analogues of cinnamoyl hydrazones were designed in such a way that, the analogues have both biological active moieties.
The structural analogues have been designed in such a way that it will show more drug likeness score than the prototype molecule but having the same pharmacophore essential for the activity. A series of fourteen N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl} eth-1-en-1-yl]benzamides [1-14] were designed and their chemical structures were drawn using Marvin sketch.
Different substituents such as electron donating (chloro, bromo, iodo), electron releasing (hydroxyl, methoxy, methyl), etc. were tried on the benzylidene ring to study their effect on the predicted properties and bioactivities.
The list of designed acyl hydrazones analogues with drug likeness scores, and other molecular properties have been listed below. The position of substituent on benzylidene ring was modified in these designed molecules to get increased drug likeness score. Table 1 shows the different substituted compounds and Table 2 indicates the nomenclature generated using software chemicalize.org.
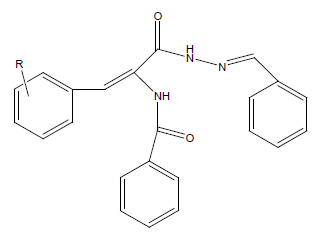
| Compound | R |
|---|---|
| 1 | H |
| 2 | 4-CH3 |
| 3 | 4-CH(CH3)2 |
| 4 | 4-OCH3 |
| 5 | 4-OH, 3-OCH3 |
| 6 | 3,5 (OCH3)2, 4 –OH |
| 7 | 3,4,5 (OCH3)3 |
| 8 | 3,5[CH(CH3)2]2, 4-OH |
| 9 | 3,4- (OCH3)2 |
| 10 | 4-OH |
| 11 | 4-Cl |
| 12 | 4-NO2 |
| 13 | 5-Br,4-OH, 3-OCH3 |
| 14 | 4-N(CH3)2 |
Table 1: Design of N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl}eth-1-en-1-yl]benzamides.
| S.No. | R | Nomenclature |
|---|---|---|
| 1 | H | IUPAC:N-[(1E)-2-phenyl-1-{N'-[(1E)-phenylmethylidene]hydrazine carbonyl}eth-1-en-1-yl]benzamide Smiles:O=C(N\N=C\C1=CC=CC=C1)C(\NC(=O)C1=CC=CC=C1)=C/C1=CC=CC=C1 InChI key:DLMPMANAKRHMHZ-HCNAOXRDSA-N |
| 2 | 4-CH3 | IUPAC:N-[(1E)-2-(4-methylphenyl)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:CC1=CC=C(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)C=C1 InChI key:BTHQODKQANHUKG-RWAPMGGUSA-N |
| 3 | 4-CH(CH3)2 | IUPAC:N-[(1E)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}-2-[4-(propan-2-yl)phenyl]eth-1-en-1-yl]benzamide Smiles:CC(C)C1=CC=C(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)C=C1 InChI key:FLFPXFZDPBDUDS-MUXKBQDCSA-N |
| 4 | 4-OCH3 | IUPAC:N-[(1E)-2-(4-methoxyphenyl)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:COC1=CC=C(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)C=C1 InChI key:DTUOCCCOSQNKIE-RWAPMGGUSA-N |
| 5 | 4-OH, 3-OCH3 | IUPAC:N-[(1E)-2-(4-hydroxy-3-methoxyphenyl)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:COC1=CC(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)=CC=C1O InChI key:FSQSWQXGEWISJE-JTDPGYMQSA-N |
| 6 | 3,5 (OCH3)2, 4 –OH | IUPAC:N-[(1E)-2-(4-hydroxy-3,5-dimethoxyphenyl)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:COC1=CC(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)=CC(OC)=C1O InChI key:LLKGXFKFKHGAMM-LZJHMVIHSA-N |
| 7 | 3,4,5 (OCH3)3 | IUPAC:N-[(1E)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}-2-(3,4,5-trimethoxyphenyl)eth-1-en-1-yl]benzamide Smiles:COC1=CC(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)=CC(OC)=C1OC InChI key:VFMVVGFCXBLVNT-DSSLOXKCSA-N |
| 8 | 3,5[CH(CH3)2]2; 4-OH | IUPAC:N-[(1E)-2-[4-hydroxy-3,5-bis(propan-2-yl)phenyl]-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:CC(C)C1=CC(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)=CC(C(C)C)=C1O InChI key:QPPLZNRJBJMEKV-CUDGALFRSA-N |
| 9 | 3,4- (OCH3)2 | IUPAC:N-[(1E)-2-(3,4-dimethoxyphenyl)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:COC1=C(OC)C=C(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)C=C1 InChI key:GFXITFWFNWTANS-WTUKZMCFSA-N |
| 10 | 4-OH | IUPAC:N-[(1E)-2-(4-hydroxyphenyl)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:OC1=CC=C(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)C=C1 InChI key:LZTMQSRUDJGABM-XMGKXQPASA-N |
| 11 | 4-Cl | IUPAC:N-[(1E)-2-(4-chlorophenyl)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:ClC1=CC=C(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)C=C1 InChI key:AQHYSIHWDFNNIY-UPXQHGRYSA-N |
| 12 | 4-NO2 | IUPAC:N-[(1E)-2-(4-nitrophenyl)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:O=C(N\N=C\C1=CC=CC=C1)C(\NC(=O)C1=CC=CC=C1)=C/C1=CC=C(C=C1)N(=O)=O InChI key:NDPLRZUHTAWBTQ-XMGKXQPASA-N |
| 13 | 5-Br, 4-OH, 3-OCH3 | IUPAC:N-[(1E)-2-(3-bromo-4-hydroxy-5-methoxyphenyl)-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:COC1=CC(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)=CC(Br)=C1O InChI key:GSVAGLOAPDWMPW-BCQUTSDVSA-N |
| 14 | 4-N(CH3)2 | IUPAC:N-[(1E)-2-[4-(dimethylamino)phenyl]-1-{N'-[(1E)-phenylmethylidene]hydrazinecarbonyl}eth-1-en-1-yl]benzamide Smiles:CN(C)C1=CC=C(\C=C(\NC(=O)C2=CC=CC=C2)C(=O)N\N=C\C2=CC=CC=C2)C=C1 InChI key:AJTCDWAWKZMUHP-KWCUAYMTSA-N |
| 15 | Diclofenac | IUPAC:2-{2-[(2,6-dichlorophenyl)amino]phenyl}acetic acid Smiles:OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl InChI key:DCOPUUMXTXDBNB-UHFFFAOYSA-N |
| 16 | Phenyl Butazone | IUPAC:4-butyl-1,2-diphenylpyrazolidine-3,5-dione Smiles:CCCCC1C(=O)N(N(C1=O)C1=CC=CC=C1)C1=CC=CC=C1 InChI key:VYMDGNCVAMGZFE-UHFFFAOYSA-N |
Table 2: Nomenclature of the designed cinnamoyl hydrazones using Chemicalize.org.
Molecular properties predictions
Lipinski’s Rule of Five describes molecular properties important for a drug’s pharmacokinetics in the human body, including their absorption, distribution, metabolism, and excretion [28]. Using Chemicalize, Mol inspiration and Mol soft molecular properties were predicted for the designed compounds. The results obtained, given in detail in Tables 3-6, revealed that all the compounds tested obeyed Lipinski rule of five with not more than 5 hydrogen bond donors (OH and NH groups), not more than 10 hydrogen bond acceptors (notably N and O), not more than 15 rotatable bonds (rotb), molecular weight under 500 g/mol and a partition coefficient, log P less than 5 except for compound 3 and 8. All the compounds tested also passed the other Lipinski like filter such as bioavailability.
| S.NO | R | FORMULA | Molecular weight | Log P | PSA | SASA |
|---|---|---|---|---|---|---|
| 1 | H | C23H19N3O2 | 369.424 | 3.99 | 70.56 | 502.18 |
| 2 | 4-CH3 | C24H21N3O2 | 383.451 | 4.50 | 70.56 | 533.75 |
| 3 | 4-CH(CH3)2 | C26H25N3O2 | 411.505 | 5.23 | 70.56 | 595.22 |
| 4 | 4-OCH3 | C24H21N3O3 | 399.45 | 3.83 | 79.79 | 550.05 |
| 5 | 4-OH, 3-OCH3 | C24H21N3O4 | 415.449 | 3.52 | 100.02 | 561.75 |
| 6 | 3,5 (OCH3)2, 4 –OH | C25H23N3O5 | 445.475 | 3.37 | 109.25 | 609.47 |
| 7 | 3,4,5 (OCH3)3 | C26H25N3O5 | 459.502 | 3.51 | 98.25 | 646.66 |
| 8 | 3,5[CH(CH3)2]2, 4-OH | C29H31N3O3 | 469.585 | 6.17 | 90.79 | 697.84 |
| 9 | 3,4- (OCH3)2 | C25H23N3O4 | 429.476 | 3.67 | 89.02 | 598.56 |
| 10 | 4-OH | C23H19N3O3 | 385.423 | 3.68 | 90.79 | 512.20 |
| 11 | 4-Cl | C23H18ClN3O2 | 403.869 | 4.59 | 70.56 | 518.47 |
| 12 | 4-NO2 | C23H18N4O4 | 414.421 | 3.93 | 116.38 | 541.90 |
| 13 | 5-Br,4-OH, 3-OCH3 | C24H20BrN3O4 | 494.345 | 4.29 | 100.02 | 581.45 |
| 14 | 4-N(CH3)2 | C25H24N4O2 | 412.493 | 4.09 | 73.80 | 589.75 |
| 15 | Diclofenac | C14H11Cl2NO2 | 296.149 | 4.26 | 49.33 | 361.15 |
| 16 | Phenyl Butazone | C19 H20 N2 O2 | 308.374 | 4.14 | 40.62 | 466.42 |
Log P- Partition co efficient, PSA- Polar surface area, SASA- Solvent Accessible Surface Area
Table 3: Molecular properties prediction of N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl}eth-1-en-1-yl]benzamides using Chemicalize.org.
| S.NO. | R | Lipinski rule of five | Bio availability | Ghose filter | Lead likeness | Mugge filter | Veber filter |
|---|---|---|---|---|---|---|---|
| 1 | H | Yes | Yes | Yes | Yes | Yes | Yes |
| 2 | 4-CH3 | Yes | Yes | Yes | No | Yes | Yes |
| 3 | 4-CH(CH3)2 | No | Yes | Yes | No | No | Yes |
| 4 | 4-OCH3 | Yes | Yes | Yes | Yes | Yes | Yes |
| 5 | 4-OH, 3-OCH3 | Yes | Yes | Yes | Yes | Yes | Yes |
| 6 | 3,5 (OCH3)2, 4 –OH | Yes | Yes | Yes | Yes | Yes | Yes |
| 7 | 3,4,5 (OCH3)3 | Yes | Yes | No | No | Yes | Yes |
| 8 | 3,5[CH(CH3)2]2; 4-OH | Yes | Yes | Yes | Yes | Yes | Yes |
| 9 | 3,4- (OCH3)2 | Yes | Yes | Yes | Yes | Yes | Yes |
| 10 | 4-OH | Yes | Yes | Yes | Yes | Yes | Yes |
| 11 | 4-Cl | Yes | Yes | Yes | No | Yes | Yes |
| 12 | 4-NO2 | Yes | Yes | Yes | Yes | Yes | Yes |
| 13 | 5-Br, 4-OH, 3-OCH3 | Yes | Yes | No | No | Yes | Yes |
| 14 | 4-N(CH3)2 | Yes | Yes | Yes | No | Yes | Yes |
| 15 | Diclofenac | Yes | Yes | Yes | Yes | Yes | Yes |
| 16 | Phenyl Butazone | Yes | Yes | Yes | Yes | Yes | Yes |
Table 4: Molecular properties prediction of N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl}eth-1-en-1-yl]benzamides using Chemicalize.org.
| Compound | nON | nOHNH | violations | nrotob | Volume(m 3) | Log P | n Atoms |
|---|---|---|---|---|---|---|---|
| 1 | 5 | 2 | 0 | 6 | 340.311 | 4.664 | 28.0 |
| 2 | 5 | 2 | 1 | 6 | 356.871 | 5.113 | 29.0 |
| 3 | 5 | 2 | 1 | 7 | 390.259 | 6.177 | 31.0 |
| 4 | 6 | 2 | 0 | 7 | 365.555 | 4.721 | 30.0 |
| 5 | 7 | 3 | 0 | 7 | 373.873 | 4.003 | 31.0 |
| 6 | 8 | 3 | 0 | 8 | 448.227 | 4.02 | 33.0 |
| 7 | 8 | 2 | 0 | 9 | 416.947 | 4.296 | 34.0 |
| 8 | 6 | 3 | 1 | 8 | 448.227 | 6.379 | 35.0 |
| 9 | 7 | 2 | 0 | 8 | 391.401 | 4.311 | 32.0 |
| 10 | 6 | 3 | 0 | 6 | 348.327 | 4.185 | 29.0 |
| 11 | 5 | 2 | 1 | 6 | 353.846 | 5.342 | 29.0 |
| 12 | 8 | 2 | 0 | 7 | 363.644 | 4.623 | 31.0 |
| 13 | 7 | 3 | 0 | 7 | 391.759 | 4.969 | 32.0 |
| 14 | 6 | 2 | 0 | 7 | 386.216 | 4.767 | 31.0 |
| Diclofenac | 3 | 2 | 0 | 4 | 241.724 | 4.337 | 19.0 |
| Phenyl Butazone | 4 | 0 | 0 | 5 | 291.939 | 4.558 | 23.0 |
nON- No of hydrogen bond acceptors, nOHNH- No of hydrogen bond donors, ntrob- No. of rotatable bonds, Log P- Partition co efficient
Table 5: Molecular properties prediction of N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl} eth-1-en-1-yl]benzamides using molinspiration.com.
| Compound | No of HBA | No of HBD | Mol Log p | Mol Log s | Mol PSA (A2) | Mol volume (A3) | No of stereo centres |
|---|---|---|---|---|---|---|---|
| 1 | 3 | 2 | 4.23 | -6.21(moles/ml) 0.12(mg/ml) | 57.48 | 379.87 | 0 |
| 2 | 3 | 2 | 4.63 | -6.67(moles/ml) 0.08(mg/ml) | 57.48 | 400.81 | 0 |
| 3 | 3 | 2 | 5.34 (>5) | -7.45(moles/ml) 0.01(mg/ml) | 57.48 | 432.93 | 0 |
| 4 | 4 | 2 | 4.32 | -6.47(moles/ml) 0.13(mg/ml) | 65.02 | 411.71 | 0 |
| 5 | 5 | 3 | 3.94 | -6.05(moles/ml) 0.37(mg/ml) | 81.66 | 423.14 | 0 |
| 6 | 6 | 3 | 3.91 | -5.34(moles/ml) 0.80(mg/ml) | 88.22 | 453.81 | 0 |
| 7 | 6 | 2 | 4.26 | -6.04(moles/ml) 0.42(mg/ml) | 80.46 | 474.98 | 0 |
| 8 | 4 | 3 | 5.96 (>5) | -8.41(moles/ml) 0.00(mg/ml) | 72.96 | 499.22 | 0 |
| 9 | 5 | 2 | 4.29 | -6.34(moles/ml) 0.19(mg/ml) | 72.74 | 443.14 | 0 |
| 10 | 4 | 3 | 3.97 | -6.07(moles/ml) 0.33(mg/ml) | 75.10 | 390.42 | 0 |
| 11 | 3 | 2 | 4.94 | -7.17(moles/ml) 0.03(mg/ml) | 57.48 | 397.06 | 0 |
| 12 | 5 | 2 | 3.90 | -7.06(moles/ml) 0.04(mg/ml) | 90.86 | 404.86 | 0 |
| 13 | 5 | 3 | 4.67 | -6.86(moles/ml) 0.07(mg/ml) | 80.59 | 443.65 | 0 |
| 14 | 3 | 2 | 4.35 | -6.54(moles/ml) 0.12(mg/ml) | 60.28 | 429.42 | 0 |
| Diclofenac | 2 | 2 | 4.06 | -3.89(moles/ml) 38.23(mg/ml) | 35.44 | 242.03 | 0 |
| Phenyl Butazone | 2 | 0 | 3.99 | -3.70(moles/ml) 62.13(mg/ml) | 34.24 | 322.97 | 0 |
Log P-Partition coefficient, HBA-Hydrogen bond acceptor, HBD-Hydrogen bond donor, PSA-polar surface area
Table 6: Molecular properties prediction of N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl}eth-1-en-1-yl]benzamides using molsoft.com.
Absorption is defined as the process involved in getting a drug from its dosage form into the body and the ability to predict the percent oral absorption is primary goal in the design, optimization and selection of potential candidates in the development of oral drugs. Topological surface area (TPSA) is another key property that has been linked to bioavailability and it was found that passively absorbed molecules with a TPSA more than 140 are thought to have low oral availability [29]. TPSA obtained for the tested compounds were below 140 indicating their good oral bioavailability. Thus these results predicted the good drug likeliness, solubility, permeability and oral bioavailability of compounds.
Drug likeness and bioactivity scores
The bioactivity scores of the above mentioned compounds were calculated for their GPCR ligand, kinase inhibitor, protease inhibitor and enzyme inhibitor activities (Table 7). For average organic molecules the probability is that if the bioactivity score is more than 0 then it is active, if -0.5 to 0 then moderately active [30]. Positive score as enzyme inhibitor was obtained for compound 6. Good kinase inhibitor and enzyme inhibition scores of the compound support the possibility of anti-inflammatory and analgesic activity [31].
| Compound | GPCR Ligand | Ion Channel Modulator | Kinase Inhibitor | Nuclear Receptor Ligand | Protease Inhibitor | Enzyme Inhibitor |
|---|---|---|---|---|---|---|
| 1 | -0.46 | -0.75 | -0.54 | -0.86 | -0.46 | -0.39 |
| 2 | -0.48 | -0.79 | -0.56 | -0.86 | -0.50 | -0.43 |
| 3 | -0.42 | -0.69 | -0.53 | -0.74 | -0.43 | -0.35 |
| 4 | -0.47 | -0.77 | -0.54 | -0.82 | -0.48 | -0.41 |
| 5 | -0.43 | -0.72 | -0.49 | -0.76 | -0.49 | -0.36 |
| 6 | 0.13 | -0.08 | 0.46 | -0.74 | -0.45 | 0.32 |
| 7 | -0.43 | -0.70 | -0.47 | -0.80 | -0.47 | -0.37 |
| 8 | -0.35 | -0.60 | -0.50 | -0.57 | -0.39 | -0.27 |
| 9 | -0.45 | -0.74 | -0.51 | -0.80 | -0.48 | -0.39 |
| 10 | -0.41 | -0.69 | -0.49 | -0.72 | -0.43 | -0.33 |
| 11 | -0.45 | -0.73 | -0.54 | -0.86 | -0.48 | -0.41 |
| 12 | -0.55 | -0.73 | -0.61 | -0.87 | --0.54 | -0.45 |
| 13 | -0.58 | -0.80 | -0.57 | -0.89 | -0.61 | -0.45 |
| 14 | -0.42 | -0.71 | -0.46 | -0.78 | -0.45 | -0.37 |
| Diclofenac | 0.04 | 0.02 | 0.33 | -0.05 | -0.06 | -0.01 |
| Phenyl Butazone | -0.01 | -0.12 | -0.30 | 0.02 | -0.02 | -0.20 |
Table 7: Bioactivity prediction of N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl}eth-1-en-1-yl]benzamides using molinspiration.com.
Drug likeness may be defined as a complex balance of various molecular properties and structure features which determine whether particular molecule is similar to the known drugs. Table 8 lists the predicted scores of selected parameters for compounds. Positive scores were obtained for the compounds indicating the potential quinazolinone nucleus and amino acid residues. Compounds 3 and 8 showed negative scores, which may be due to the molecular properties that do not obey the Lipinski rule of five.
| Compound | Drug Likeness score |
|---|---|
| 1 | 0.19 |
| 2 | 0.11 |
| 3 | -0.21 |
| 4 | 0.42 |
| 5 | 0.51 |
| 6 | 0.95 |
| 7 | 0.27 |
| 8 | -0.19 |
| 9 | 0.26 |
| 10 | 0.68 |
| 11 | 0.25 |
| 12 | 0.80 |
| 13 | 0.21 |
| 14 | -0.11 |
| Diclofenac | 0.86 |
| Phenyl Butazone | 0.94 |
Table 8: Drug likeness score prediction of N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl}eth-1-en-1-yl]benzamides using molsoft.com.
Conventional non-steroidal anti-inflammatory drugs are profoundly used in the treatment of wide variety of inflammatory conditions and they act by the inhibition of cyclooxygenase (COX), the enzyme involved in the biosynthesis of prostaglandins, prostacyclins and thromboxanes from arachidonic acid [32].
Selective inhibition of COX-2 has been proposed as an approach to reduce their associated side effects while maintaining efficacy. The improved safety profile of selective COX-2 inhibitors will allow more widespread and sustained use than is currently possible with standard NSAIDs [33].
The predicted molecular properties obeyed Lipinski’ rule of five, good drug likeness scores and bioactivity scores were also obtained with these compounds. So, docking studies were performed on Compounds 3, 4, 5, 6, 7, 8, 9, 10, 13 and 14 with COX-2 enzyme (PDB ID: 6COX, resolution 2.5) and compared with reference ligand, SC-558, a selective COX-2 inhibitor. Table 9 lists the binding energies obtained.
| Compound | Estimated ΔG(Kcal/mol) a |
|---|---|
| 3 | -5.1 |
| 4 | -6.4 |
| 5 | -7.5 |
| 6 | -8.3 |
| 8 | -5.8 |
| 9 | -5.5 |
| 10 | -7.4 |
| 13 | -3.8 |
| 14 | -4.3 |
| SC-558 | -8.4 |
| Phenyl butazone | -9.2 |
Table 9: Docking studies on selected synthesized compounds with Target COX-2 (PDB ID: 6-COX).
SC-558, a diaryl heterocyclic inhibitor has 1900-fold selectivity for COX-2 over COX-1. It has a central pyrazole ring and a sulphonamide substituent attached to one of the aryl rings. The interactions observed with SC-558 at the active site of the coenzyme COX-2 were as following, the bromophenyl ring was bound in a hydrophobic cavity formed by Trp356B, Tyr 354B, Phe350B, Ser499B and Leu500B. The trifluoromethyl group was bound pocket formed by Val 318B, Leu 321B and Arg89B and. The sulphonamide group extended into an adjacent relatively polar region and interacted with Val 492B, Phe487B,Gln161B, His58B and Arg482B. The central pyrazole ring showed strong interactions with Tyr324B, Val318B and Ala496B. These features of SC-558 binding are quite important in studying the binding modes of various selective and non-selective COX-2 inhibitors.
Further the docking studies on standard drug phenyl butazone showed similar interactions at the active site of COX-2. The phenyl ring was found in the hydrophobic pocket formed by Tyr324B, Ser322B, His58B, Arg482B and Phe487B. The other phenyl ring at position of the quinazolinone ring was bound in an adjacent pocket formed by Trp356B and Tyr354B. The butyl group extended into a relatively hydrophobic region and interacted with Leucine500B, Arg89b, Leu328B and Val85B. Oxygen of carbonyl groups interacted with Arg 89B and Ser499B.
Compound 6 exhibited strongest interactions as observed with the substituted phenyl ring, it was found in a hydrophobic cavity formed by Trp356B, Tyr 354B, Phe350B, Ser499B and Leu500B and 4-hydroxy group is showing hydrogen bonding with OH of Tyr 354B (~ 2.469 Ǻ) and the oxygen of methoxy group with OH of Ser499B (~ 2.127 Ǻ). The hydrazone scaffold extended into the region containing Val318B, Ala 496B, Leu 500B, Arg89B and Leu328B. The amide -NH showed good interaction with Leu321B and –C=O showed good interaction with Ser322B. The phenyl ring of benzoyl group was found in the hydrophobic pocket formed by Tyr324B, Ser322B, His58B, Arg482B and Phe487B.
Compound 5 exhibited similar type of interactions, with the strongest interaction of 4-hydroxy group with OH of Tyr 354B. Phenyl ring was found in a hydrophobic cavity formed by Trp356B, Tyr 354B, Phe350B, Ser499B and Leu500B and the hydrazone scaffold interacted with Val318B, Ala 496B, Leu 500B and Arg89B. The hydrophobic pocket formed by Tyr324B, Ser322B, His58B, Arg482B and Phe487B was occupied by the benzoyl group. Figures 1-3 clearly indicate the similarity in the interactions, observed, in which superimposition of the tested compounds, standard drug and reference ligands can be seen.

Figure 1: Superimposition of compound 6 (golden coloured ball and stick model) and phenyl butazone (green coloured ball and stick model) and hypothetical binding interactions with amino acids (yellow coloured wire model) in the COX-2 active site (PDB ID: 6COX).

Figure 2: Compound 6 and phenyl butazone in the hydrophobic pockets of the COX-2 active site (PDB ID: 6COX).

Figure 3: Superimposition of compound 6 (blue stick model), SC558 (silver stick model) and phenyl butazone (purple coloured stick model) and hypothetical interactions with amino acids (violet colour wire model) in the COX-2 active site (PDB ID: 6COX).
The compounds which did not exhibit good anti-inflammatory activity showed poor docking interactions with large variations in their binding energies. The compound 14 exhibited interactions extended to other amino acids and different pockets other than to those observed with SC-558 and phenyl butazone which can be seen clearly in Figures 4 and 5.

Figure 4: Superimposition of compound 5 (violet ball and stick model), SC558 (blue stick model) and phenyl butazone (violet coloured stick model) and hypothetical interactions with amino acids (orange colour wire model) in the COX-2 active site (PDB ID: 6COX).

Figure 5: Superimposition of compound 14 (green ball and stick model), SC558 (light green stick model) and phenyl butazone (grey coloured stick model) and hypothetical interactions with amino acids (purple wire model) in the COX-2 active site (PDB ID: 6COX) showing that compound 14 (green ball and stick model) moved far away from the active site.
The binding interactions observed in the molecular docking studies suggest the possible COX-2(PDB ID: 6COX) inhibitory activity with compound 6. The conformational orientation observed with the selected compound possessing electron rich functionalities exhibited important
interactions pertaining to hydrogen bonding with amino acids in the relatively polar region, van der Waal’s interactions and hydrophobic interactions of the aromatic groups which found embedded into the hydrophobic cavities of the target proteins.
Conclusion
Computer based drug design on the basis of SAR and pharmacophores has become a useful tool for modern structurebased rational drug design .Inspired by the literature survey about the potentiality of acylhydrazone derivatives it was planned to design a series of N-[(1E)-2-substitutedphenyl-1-{N'-[(1E)-phenyl methylidene] hydrazine carbonyl}eth-1-en-1-yl]benzamides. Compound 6 showed similar binding interactions as that of standard drugs, Phenyl butazone and SC-5 at the binding sites of COX-2 suggest the possible COX- 2 inhibition mechanism. In silico evaluation including molecular properties like bioavailability, Lipinski’s rule of five reflected the oral bioavailability of these compounds. Good drug likeness and bioactivity scores further indicate the probable potentiality of these compounds as future drugs.
References
- Ertl P, Mühlbacher J, Rohde B, Selzer P (2003) Web-based cheminformatics and molecular property prediction tools supporting drug design and development at Novartis. SAR QSAR Environ Res 14: 321-328.
- John Wiley & Sons (1995) Kirk-OthmerEncyclopaedia Chemical Technology. 4th Edition, Volume 13.
- Abdelrazek FM, Metz P, Metwally NH, El-Mahrouky SF (2006) Synthesis and molluscicidal activity of new cinnoline and pyrano [3-c]pyrazole derivatives. Arch Pharm (Weinheim) 339: 456-460.
- Friedman JF, Mital P, Kanzaria HK, Olds GR, Kurtis JD (2007) Schistosomiasis and pregnancy. Trends Parasitol 23: 159-164.
- Sova M (2012) Antioxidant and antimicrobial activities of cinnamic acid derivatives. Mini Rev Med Chem 12: 749-767.
- Bernini R, Mincione E, Barontini M, Provenzano G, Setti L (2007) Obtaining 4-vinylphenols by decarboxylation of natural 4-hydroxycinnamic acids under microwave irradiation. Tetrahedron 63: 9663–9667.
- Chung HS, Shin JC (2007) Characterization of antioxidant alkaloids and phenolic acids from anthocyanin-pigmented rice (Oryza sativa cv. Heugjinjubyeo). Food Chem 104: 1670–1677.
- Bezerra DP, Castro FO, Alves AP, Pessoa C, Moraes MO, et al. (2006) In vivo growth-inhibition of Sarcoma 180 by piplartine and piperine, two alkaloid amides from Piper. Braz J Med Biol Res 39: 801-807.
- Naz S, Ahmad S, AjazRasool S, AsadSayeed S, Siddiqi R (2006) Antibacterial activity directed isolation of compounds from Onosmahispidum. Microbiol Res 161: 43-48.
- Nardini M, D'Aquino M, Tomassi G, Gentili V, Di Felice M, et al. (1995) Inhibition of human low-density lipoprotein oxidation by caffeic acid and other hydroxycinnamic acid derivatives. Free RadicBiol Med 19: 541-552.
- Koshihara Y, Neichi T, Murota S, Lao A, Fujimoto Y, et al. (1984) Caffeic acid is a selective inhibitor for leukotriene biosynthesis. BiochimBiophysActa 792: 92-97.
- Rouzer CA, Marnett LJ (2009) Cyclooxygenases: Structural and functional insights. J Lipid Res 50 Suppl: S29-34.
- Pontiki E, Hadjipavlou-Litina D (2006) Antioxidant and anti-inflammatory activity of aryl-acetic and hydroxamic acids as novel lipoxygenase inhibitors. Med Chem 2: 251-264.
- Pontiki E, Hadjipavlou-Litina D (2007) Synthesis and pharmacochemical evaluation of novel aryl-aceticacid inhibitors of lipoxygenase, antioxidants and anti-inflammatory agents. Bioorg Med Chem 15: 5819–5827.
- Pontiki E, Hadjipavlou-Litina D, Geromichalos G, Papageorgiou A (2009) Anticancer activity and quantitative-structure activity relationship (QSAR) studies of a series of antioxidant/anti-inflammatory aryl-acetic and hydroxamic acids. ChemBiol Drug Des 74: 266-275.
- Pontiki E, Hadjipavlou-Litina D, Litinas K, Nicolotti O, Carotti A (2011) Design, synthesis and pharmacobiological evaluation of novel acrylic acid derivatives acting as lipoxygenase and cyclooxygenase-1 inhibitors with antioxidant and anti-inflammatory activities. Eur J Med Chem 46: 191-200.
- Pontiki E, Hadjipavlou-Litina D, Litinas K,Geromichalos G (2014) Novel cinnamic acid derivatives as antioxidant and anticancer agents: Design, synthesis and modeling studies. Molecules 19: 9655-9674.
- Pontiki E, Hadjipavlou-Litina D (2008) Lipoxygenase inhibitors: A comparative QSAR study review and evaluation of new QSARs. Med Res Rev 28: 39-117.
- Morphy R, Kay C, Rankovic Z (2004) From magic bullets to designed multiple ligands. Drug Discov Today 9: 641-651.
- Morphy R, Rankovic Z (2005) Designed multiple ligands. An emerging drug discovery paradigm. J Med Chem 48: 6523-6543.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46: 3-26.
- Chen G, Zheng S, Luo X, Shen J, Zhu W, et al. (2005) Focused combinatorial library design based on structural diversity, druglikeness and binding affinity score. J Comb Chem 7: 398–406.
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, et al. (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45: 2615-2623.
- Lalitha P, Sivakamasundari S (2010) Calculation of molecular lipophilicity and drug likeness for few heterocycles, Oriental J Chem 26: 135-141.
- Verma A (2012) Lead finding from Phyllanthusdebelis with hepatoprotective potentials. Asian Pacific J Trop Biomed 1735-S1737.
- Nirmala H, Mullaicharam AR (2012) Molecular modifications of ibuprofen using in silico modeling system. Int J NutrPharmacolNeur Diseases 2: 156-162.
- Warner TD, Mitchell JA (2004) Cyclooxygenases: New forms, new inhibitors and lessons from the clinic. FASEB J 18: 790-804.
- Mosaad Mohamed S, Mohseen Kamal M, EmadKassem MM, NagehAbotalebKhedr M (2011) Synthesis, biological evaluation and molecular docking of quinazoline-4(1H)-one derivatives as anti-inflammatory and analgesic agents. Act Pol Pharm Drug Res 68: 665-675.
Citation: Banu BH, Rajitha G, Bharathi K (2016) An Approach to Computer Aided Drug Design of some Bioactive Cinnamoyl Hydrazones, In Silico and Docking Studies as Possible COX-2 Selective Inhibitors. J Biotechnol Biomater 6:240. DOI: 10.4172/2155-952X.1000240
Copyright: © 2016 Banu BH, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 12244
- [From(publication date): 9-2016 - Aug 20, 2025]
- Breakdown by view type
- HTML page views: 11248
- PDF downloads: 996