Immunoreactivity of Anti-AβP-42 Specific Antibody with Toxic Chemicals and Food Antigens
Received: 04-May-2018 / Accepted Date: 02-Jun-2018 / Published Date: 09-Jun-2018 DOI: 10.4172/2161-0460.1000441
Keywords: AβP-42; Amyloidogenesis; Alzheimer’s Disease; Toxins; Dietary Proteins; Immunoreactivity; Neurodegeneration
Abbrevations
OD: Once in a Day; RT: Room Temparature.
Introduction
According to the National Institute on Aging, Alzheimer’s disease (AD) is the sixth leading cause of death in the United States [1]. In most people, the exact causes of AD are not fully understood. In cases of early onset of AD, genetic component plays a significant role. Late-onset Alzheimer’s, the most common form of the disease, is thought to arise from a combination of genetic, environmental and lifestyle factors that may cause a series of brain changes to occur over decades [1]. The genetic component of AD and its association with APOE-ε4 is well established [2]. Apolipoprotein E (ApoE) is a major cholesterol carrier that supports lipid transport and injury repair in the brain; the ApoE-ε4 allele is not effective as the other variants, so much so that it is the largest known genetic risk factor for late-onset sporadic AD [3]. The APOE-ε4 allele is also associated with increased risk for cerebral amyloid angiopathy and age-related cognitive decline during normal ageing [4].
In relation to the role of environmental triggers, many studies have established that specific infectious agents such as Herpes simplex virus type 1 (HSV-1) [5,6], Gram-negative bacteria [7], Chlamydia pneumoniae [5,8], several types of spirochetes including Borrelia burgdorferi [5,9], fungal infections [5,10,11] oral pathogens [12-16], and bacterial toxins [17,18] are implicated in AD. However, with the exception of a few chemicals, the role of environmental toxins and dietary proteins in association with AD has largely been neglected [19]. Toxic metals such as aluminum [20-24], mercury [25-27] and lead [28, 29] are among the few that are known to cause toxicity to the brain and other organs and have been linked to numerous neurodegenerative disorders, including AD.
Exposure to aluminum and such metals has been followed by aggregation of amyloid-β protein (AβP) on neuronal cells [28] as well as AD-like pathologies, which have been shown in animals as well as in human subjects [29]. Aluminum-induced neurotoxicity includes oxidative stress, mitochondrial dysfunction, inflammatory response, and neurofibrillary degeneration, possibly through amyloid-β oligomerization [20,22]. In vitro studies have shown that aluminum together with other metals is involved in the formation of AβP aggregation, which leads to amyloid fibrils and the formation of amyloidlike plaque structure [24]. As early as 1999 aluminum had already been shown to be neurotoxic, causing abnormal clustering and aggregation that resulted in neuronal death [30]. A review in 1998 [31] showed that such accumulations of amyloid and extracellular tangles act as irritants, resulting in inflammatory reactions that lead to the production of potentially neurotoxic products that contribute to neuronal loss.
Mercury has also been reported as a risk factor for AD due to its well-known neurotoxicity. Mercury ions bind to tubulin, inhibiting guanosine triphosphate (GTP) nucleotide binding capacity and reducing its biological activity, leading to microtubule degeneration [25]. In vitro and animal studies have shown that mercury causes hyperphosphorylation of tau protein and increased formation of AβP aggregation [26].
Phthalates and bisphenol A (BPA) are used as plasticizers in water bottles, food cans, and many other products. As such, they can leach or migrate into food and water, and hence through estrogenic activity or epigenetic modification may affect human health [32,33]. If BPA crosses the blood-brain barrier, it can bind to a target enzyme called protein disulfide isomerase (PDI). PDI is a stress protein found in the endoplasmic reticulum of many cells, including neural tissue, and is involved in protein folding. Normally this enzyme effectively inhibits α-synuclein fibril formation, but the S-nitrosylation of PDI by chemicals leads to a loss of enzymatic activity and the enhancement of protein misfolding and α-synuclein aggregation that are found in AD and Parkinson’s disease [34,35]. It is important to note that BPA has been shown to affect the prefrontal cortex and hippocampus where it interferes with synapse formation, resembling events that occur in AD [36,37].
Prenatal exposure to phthalates was shown to be associated with poor cognition and social impairment mainly in girls, who are more vulnerable to the neurotoxic effects of phthalates than boys [38-40]. Phthalates have also been shown to significantly inhibit the activity of acetylcholinesterase, and upregulate myelin basic protein (MBP) and glial fibrillary acidic protein (GFAP) in a zebra fish model [41]. Furthermore, it was observed that prenatal exposure to phthalates caused cognitive dysfunction and an increase in tau protein phosphorylation in rats [42,43]. Organic solvents containing benzene ring, such as phenol and alkylphenol, are found in the paint, paper, cleansing agent, and textile industries, and have been shown to exert an estrogenic effect and an increase in the expression of amyloid and precursor protein-2 accumulation, which may result in neuronal degeneration in AD brains [19,44]. Since humans are exposed to so many chemicals, their synergistic effect may contribute to immunotoxic, neurotoxic and AD-like pathology [19,45,46]. Autoantibodies to AβP are detected in the elderly and in AD. These antibodies may exert a protective role by inhibiting the activity of toxic peptides. They may also induce an immune attack against AβP, activating inflammatory cascades that kill the neurons in which AβP resides [47,48]. The exact source of these antibodies is not clear. They could be derived as a response to haptenic chemicals bound to AβP or other proteins or peptides, or these antibodies may also arise from a reaction to other antigens, such as pathogens or foods that cross-react with the amyloid peptides [49-52]. A search for food proteins that share similarity with AβP was done by Carter [49], who found that many foods display a large number of tetrapeptide sequences matching those of β-amyloid. This suggests that these and possibly other food proteins could play a rather unexpected role in AD. We resolved then to examine whether this close homology of diverse antigens with AβP and possibly the misfolding of proteins generated by the binding of haptenic chemicals to various proteins constitute an autoimmune component of AD that is triggered by these homologies and neo-antigen formation. To confirm this immunoreactivity or cross-reactivity, we applied monoclonal antibodies made against AβP-42 with 20 haptenic chemicals bound to human serum albumin (HSA) as well as with 208 purified dietary proteins commonly consumed in raw and cooked forms.
Materials and Methods
Monoclonal antibodies
Commercially available antibodies were purchased from different companies. Rabbit monoclonalanti-amyloid-β1-42(fibrilsequence DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA) produced by Abcam’s RabMab® technology was purchased from Abcam. This antibody reacts strongly to human Aβ 42 monomers, oligomers and fibrils, but not with human muscle fibrils. Additional information about the specificity of this antibody is provided in the Abcam package insert (ab201061) and in an article by Hatami et al. [53]. Affinity-purified mouse anti-amyloid-β1-42 was purchased from BioLegend, San Diego, CA USA. This antibody reacted strongly with formalin-fixed, paraffin- embedded diseased human brain tissue.
The mouse monoclonal antibody (mA β P2-1) to AβPP peptide residues of 104-118 CKTHPHFVIPYRCL was purchased from Thermo Fisher Scientific. Its specificity was confirmed by Western blot, immunoprecipitation, ELISA, and immunohistochemistry. According to the product package insert, this antibody is specific for native, nondenatured amyloid precursor protein (APP) from human and monkey. It does not cross-react with mouse, rat APP or other APP homologs.
Proteins and peptides
Amyloid β -peptide 1-42 at purity greater than 95% was purchased from GenScript Piscataway, NJ, USA. Recombinant tau protein was obtained from rPeptide Watkinsville, GA USA. Wheat globulin, β + β casein, glutamic acid decarboxylase peptide was synthesized by BioSynthesis Lewisville, TX USA.
Chemicals bound to HSA
Dinitrophenyl-HSA (DNP-HSA), aflatoxin-HSA, diethyl phthalate, and other chemicals were purchased from Sigma-Aldrich, St. Louis, MO, USA. Protein disulfide isomerase (PDI) was purchased from Creative Biomart.
Binding of phthalate to HAS
Preparation of diethyl phthalate was done according to the method described by Zhou et al. [54]. Briefly, 2 mg of diethyl phthalate was dissolved in methanol and added dropwise to 25 mg of HSA dissolved in 5 mL of 0.14 Tris-HCl buffer. After stirring for 1 hour at room temperature, the conjugates were dialyzed using tubes at a cutoff of 6,000 Dalton against four changes of 0.01 M PBS pH 7.2. After completion of dialysis, the mixture was filtered and kept at -20°C until used.
Binding of mercury to HAS
For this preparation, 100 mg of HSA was dissolved in 9 mL of buffer solution containing potassium chloride and sodium borate 0.05 M, and the pH was adjusted to 9.4 with 0.1 N NaOH. Then, 25 mg of mercury chloride was dissolved in 1 mL of buffer and added dropwise to the HSA solution. The reaction mixture was stirred overnight, dialyzed against 0.1 M PBS using tubing with a cutoff of 6,000 Da kept at -20°C.
Binding of aluminum and glyphosate to HAS
Aluminum hydroxide bound to HSA was prepared according to Lu et al. [55]. Glyphosate or N-phosphomethylglycine was bound to HSA using the method described by Yue et al. [56].
All other chemicals bound to HSA including pyrethroid-HSA and formaldehyde-HSA were done according to the methodology described by Vojdani in 2014 [57].
Preparation of dietary antigens
Food antigens were prepared from products purchased from the supermarket in both raw and cooked forms. For that preparation, 10 g of food product was put in a food processor using 0.1 M of phosphate buffer saline (PBS) at pH 7.4. The mixer was turned on and off for 1 h and then kept on the stirrer overnight at 4°C. After centrifugation at 20,000 g for 15 min, the top layer, which contained oil bodies, was discarded. The liquid phase was removed and dialyzed against b0.01 M of PBS using dialysis bags, with a cutoff of 6,000 kDa. Dialysis was repeated three times to ensure all small molecules were removed. After dialysis, all samples were filtered through a 0.2 micron filter to remove any debris. Protein concentrations were measured using a kit provided by Bio-Rad (Hercules, CA, USA). Different peptides were purchased from Bio-Synthesis (Lewisville, TX, USA). Lectin and agglutinins including pea lectin and lentil lectin were purchased from Sigma- Aldrich (St. Louis, MO, USA). The complete list of foods used for antigen preparation is shown in [58].
Enzyme-linked immunosorbent assay (ELISA)
For demonstration of anti-AbP-42 antibody binding to chemicals bound to HSA and food antigens. Chemicals bound to HSA, food antigens, PDI enzyme, and AβP-42 were dissolved in PBS or methanol at a concentration of 1.0 mg/mL and then diluted 1:100 in 0.1 M carbonate-bicarbonate buffer at a pH of 9.5, and 100 μL was added to each well of the polystyrene flat-bottom ELISA plate. Plates were incubated overnight at 4°C and then washed three times with 200 μL 0.01 M PBS containing 0.05% Tween 20 at a pH of 7.4. The non-specific binding of immunoglobulins was prevented by adding 2% bovine serum albumin (BSA) into the PBS and then incubating overnight at 4°C.
Plates were washed, and then, rabbit monoclonal antibodies diluted at an optimal dilution of 1:500 were added to quadruplicate wells coated with each antigen. Plates were incubated for an additional 1 h at room temperature. The plates were then washed five times with Tris-buffered saline (TBS)-Tween. Alkaline phosphatase-labeled secondary antibody at a dilution of 1:600 was then added to all wells and incubated again for 1 hour at room temperature. The enzyme reaction was started by adding 100 μL of paranitrophenylphosphate at a concentration of 1 mg/mL in diethanolamine buffer containing 1 mM MgCl2 and sodium azide at a pH of 9.8. The reaction was stopped 45 min later with 50 μL of 1 N NaOH, and the samples were read by an ELISA reader; the optical densities were recorded.
To determine the specificity of affinity-purified rabbit anti-AβP-42 binding to the chemicals bound to HSA and food antigens, the rabbit anti-AβP-42 was replaced with the same dilution of non-immunized rabbit serum and added to quadruplicate wells. Furthermore, the anti- AβP-42 and other reagents were added to 4 wells coated with HSA and 4 wells coated with 2% BSA alone; these were then used as negative controls. After the addition of other ELISA reagents to these 12 control wells, the ODs were measured and their mean was subtracted from the mean OD of all other reactions.
Demonstration of specificity of anti-AβP-42 binding to chemicals-HSA and food antigens
To determine the specificity of this antigen-antibody reaction, serial dilutions of serum as well as inhibition studies were conducted. Anti- AβP-42 was serially diluted from 1:400-1:12,800 in PBS containing 2% BSA, and was then applied to different wells of ELISA plates coated with DNP-HSA, phthalate-HSA, egg yolk and cooked tuna antigens. After the completion of all other ELISA steps, the recorded ODs were converted to different graphs.
Inhibition study
Monoclonal rabbit anti-AβP-42 in the presence or absence of chemicals-HSA or food antigens was used in the inhibition study. In different test tubes, 1 mL of 1:400 diluted rabbit anti-AβP-42 was preincubated with 100 μL of diluent containing 100 μg of BSA, HSA, or 1.5-100 μg each of DNP-HSA, phthalate-HSA, egg yolk antigen or cooked tuna antigen. After mixing, the tubes were kept for 1 hour in a 37°C water bath, followed by 4 hours of incubation at 4°C, and then centrifugation at 3,000 g for 10 min. The supernatant was used for measuring the degree of anti-AβP-42 binding to the chemical-HSA or food antigen- coated plates before and after absorption with the specific antigens. After completion of the ELISA procedure, the ODs were converted into graphs.
Measurement of AβP-42 concentrations by Sandwich ELISA
For measurement of AβP-42 concentrations, 100 μL of affinitypurified mouse anti-AβP-42 at a concentration of 500 μg/mL and a dilution of 1:200 in 0.1 M carbonate buffer pH 9.6 were applied to the surfaces of the wells of microtiter ELISA plates. After incubation, addition of 2% BSA, and washing, AβP-42 in standard concentrations of 40, 80, 160, 320, 640, 960, and 1280 pg/mL was prepared, and 100 μL of each preparation were added to the first row of the microtiter plate coated with the antibody. Also, known amounts of AβP-42 in concentrations of 31.25, 62.5, 125, 250, 500 and 1000 pg/mL each were added to duplicate wells. Furthermore, tau protein, α+β casein, and glutamic acid decarboxylase peptides in concentrations of 31.2 to 1000 pg/mL were added to the additional duplicate wells coated with anti- AβP-42 antibody. After 60 min incubation at RT and washing, 100 μL of rabbit monoclonal anti-AβP-42 antibody were added to all wells.
The binding of pure Aβ-42 peptide and other proteins to the Aβ-42 first antibody and its sandwich with the second antibody was measured by the addition of alkaline-phosphatase labeled anti-rabbit, followed by substrate and color development, then reading of the ODs at 405 nM. After generation of a standard curve, the obtained values of AβP-42 concentration were compared to the true values.
Results
Using rabbit monoclonal antibody against AβP-42, we measured immune reactivity with various chemicals bound to HSA and compared them with the level of this immune reaction with its target antigen AβP- 42 as a positive control, and HSA alone as a negative control.
Compared to the mean OD of control wells coated with HSA alone or an OD of 0.17, the anti- AβP-42 specific antibody reacted with a phthalate-HSA OD of 1.9, aluminum-HSA OD of 1.2, DNPHSA OD of 1.1, and reacted to a lower degree with a mercury- HSA OD of 0.75. Compared to 3SD above the mean of the ODs of the control wells, the p values for phthalate-HSA, aluminum-HSA, DNP-HSA, and mercury-HSA were p<0.00001. Reactions of the same antibody with aflatoxin-HSA, glyphosate-HSA, and pyrethroid-HSA were similar to the control wells (Figure 1). Furthermore, the reaction of this antibody with formaldehyde-HSA, isocyanate-HSA, bisphenol A, tetrebromobisphenol A, tetrachloroethylene, and parabens was also comparable to the wells coated with HSA alone (data not shown). Since PDI is a stress protein that is involved in the inhibition of protein misfolding and in the aggregation of α- synuclein, the reactivity of AβP-42 antibody with PDI was examined. The reaction of anti- AβP- 42 antibody with PDI resulted in an OD of 1.0 (23%), which is considered moderate (Figure 1). Similarly, by testing the anti-AβP-42 antibody for its possible immunoreactivity with 208 different food antigens, we found that in comparison to AβP-42 antibody binding with AβP-42 peptide with an OD of 3.8, numerous foods reacted from moderately to strongly with this antibody. The OD for wells coated with BSA alone was 0.15, for spinach 0.32, and for egg white 0.45, which were slightly greater than 3SD above the mean of control wells. This immunoreactivity was significantly higher (p<0.00001) with seaweed and scallop with an OD of 0.6, cashew vicilin with an OD of 0.8, wheat extract and pea lectin with an OD of 0.85, pea protein with an OD of 1.4, egg yolk with an OD of 1.55, cow’s milk with an OD of 1.8, α+β casein with an OD of 2.4, and for α-gliadin 33-mer and other wheat peptides such as β-amylase, CXCR3-binding gliadin peptide, and wheat globulin with ODs from 2.4 to 3.2 (62-83% of anti- AβP binding to AβP-42. In addition to milk and wheat antigens, the strongest reactions of AβP- 42 antibody was observed with canned tuna extract (OD of 2.8) but not raw tuna (only a moderate OD of 0.7), lentil lectin (OD of 2.1), squid (OD of 2.1), and hazelnut (OD of 2.0) (Figure 2). The reactivity of this antibody with the additional 189 food extracts used in this assay was comparable to the wells coated with BSA alone (Figure 1).
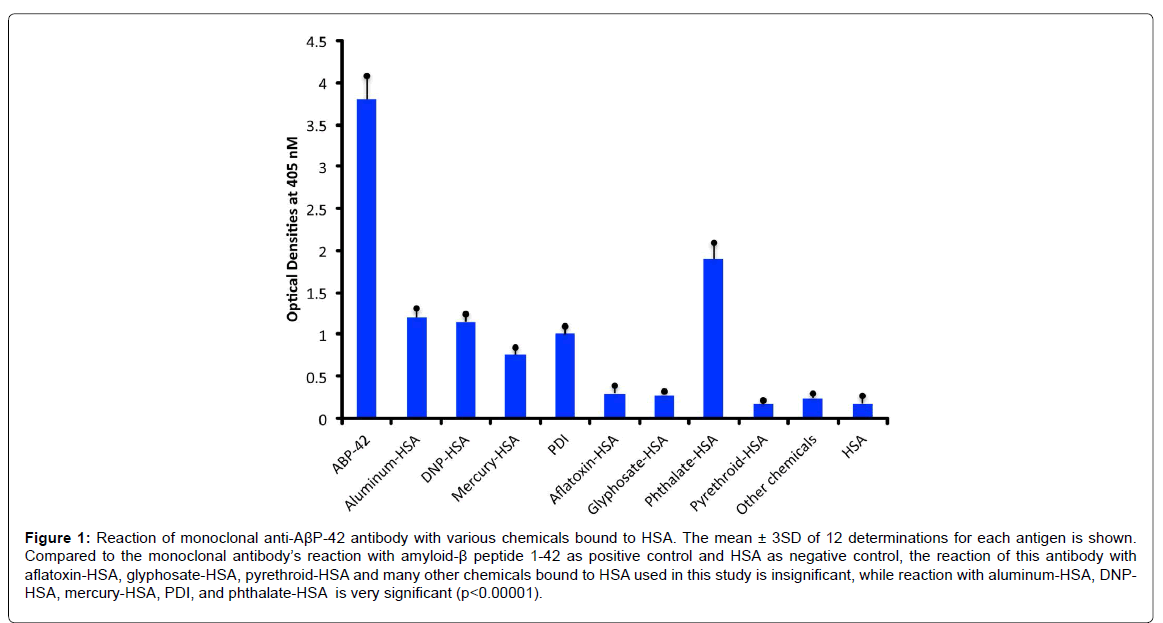
Figure 1:Reaction of monoclonal anti-AßP-42 antibody with various chemicals bound to HSA. The mean ± 3SD of 12 determinations for each antigen is shown. Compared to the monoclonal antibody’s reaction with amyloid-ß peptide 1-42 as positive control and HSA as negative control, the reaction of this antibody with aflatoxin-HSA, glyphosate-HSA, pyrethroid-HSA and many other chemicals bound to HSA used in this study is insignificant, while reaction with aluminum-HSA, DNPHSA, mercury-HSA, PDI, and phthalate-HSA is very significant (p<0.00001).
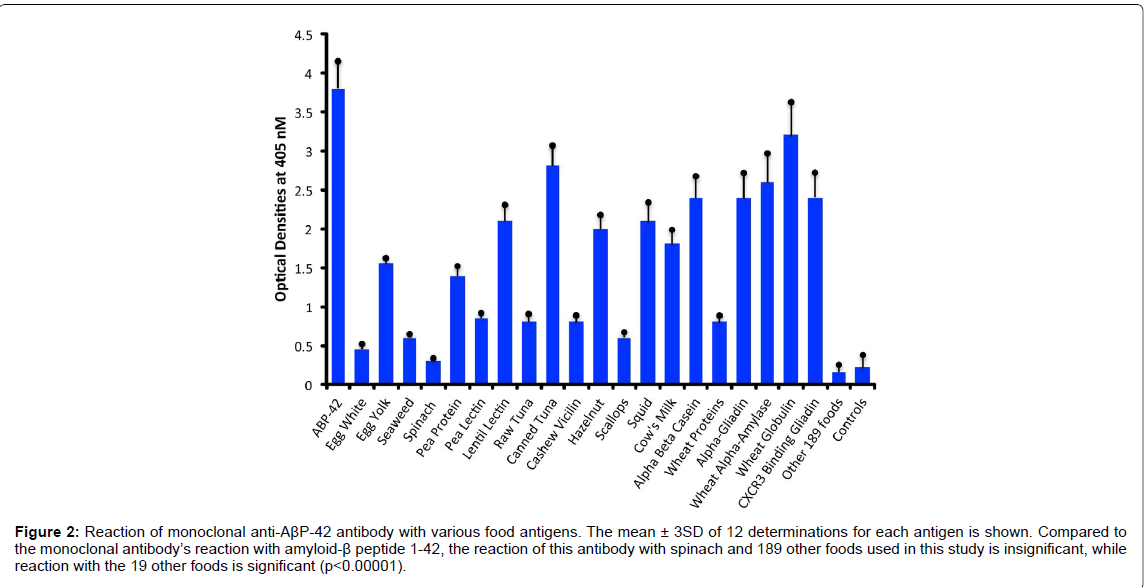
Figure 2:Reaction of monoclonal anti-AßP-42 antibody with various food antigens. The mean ± 3SD of 12 determinations for each antigen is shown. Compared to the monoclonal antibody’s reaction with amyloid-ß peptide 1-42, the reaction of this antibody with spinach and 189 other foods used in this study is insignificant, while reaction with the 19 other foods is significant (p<0.00001).
Determination of the specificity of anti-AβP-42 antibody binding to chemicals-HSA and food antigens
First, we used unimmunized rabbit serum and examined its reaction with chemicals bound to HSA and with all 208 food antigens, obtaining no OD above 0.25. We also used mouse monoclonal antibody made against AβP 104-118 AA, and reacted them with all chemicals bound to HSA or the 208 foods. None of the ODs were higher than 0.3. These results support the specificity of AβP-42 binding to some of the chemicals bound to HSA or food antigens listed in (Figures 1 and 2). Further experiments such as dilutions of anti-AβP-42 antibody and inhibition of the antibody-antigen reaction were performed (Figure 2).
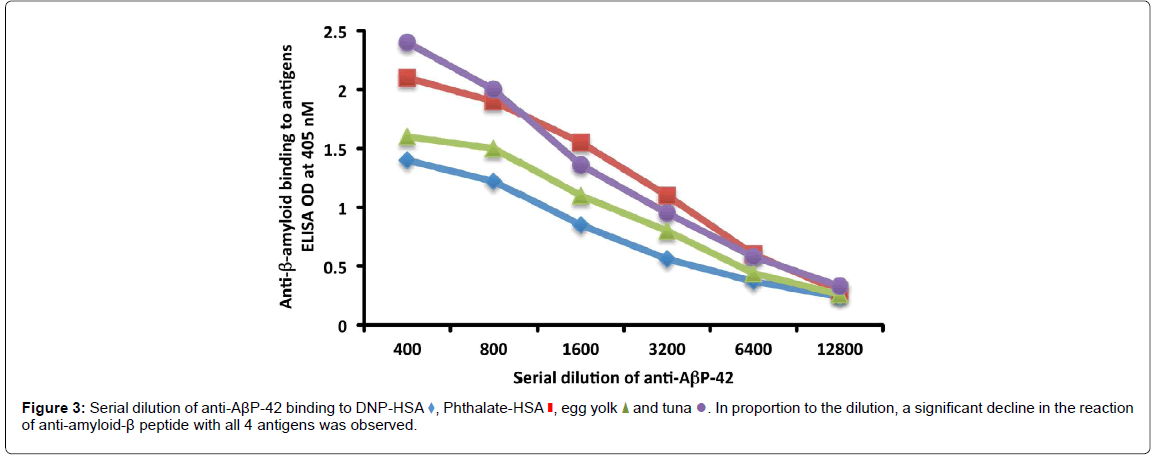
Figure 3:Serial dilution of anti-AßP-42 binding to DNP-HSA , Phthalate-HSA
, Phthalate-HSA  , egg yolk
, egg yolk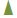 and tuna
and tuna  . In proportion to the dilution, a significant decline in the reaction of anti-amyloid-ß peptide with all 4 antigens was observed.
. In proportion to the dilution, a significant decline in the reaction of anti-amyloid-ß peptide with all 4 antigens was observed.
As shown in (Figure 3), in proportion to the dilutions of anti- AβP-42 antibody, the ODs of the antibody binding to DNP-HSA, phthalate-HSA, egg yolk antigen or tuna antigen declined significantly. For example, the reaction of anti-AβP-42 at a dilution of 1:400 with tuna antigen gave an OD of 2.4, a dilution of 1:3200 gave an OD of 0.95, and a dilution of 1:12800 gave an OD of 0.33, which is almost equivalent to the ELISA background (Figure 3).
To further demonstrate the specificity of this antibody-antigen reaction, different amounts of chemicals bound to HSA or food proteins (inhibitors) in concentrations ranging from 3-100 μg were added to the antibody before performing the ELISA on the plates coated with the optimal concentrations of the same antigens. The addition of the anti- AβP-42 antibody in the presence of higher concentrations of specific antigens in the liquid phase resulted in significant inhibition of the anti- AβP-42 antibody binding to the chemicals-HSA or food antigens. This inhibition of antigen-antibody reaction was insignificant when HSA was added to the liquid phase or when the antigen concentration in the liquid phase was below 6 μg (Figure 4).
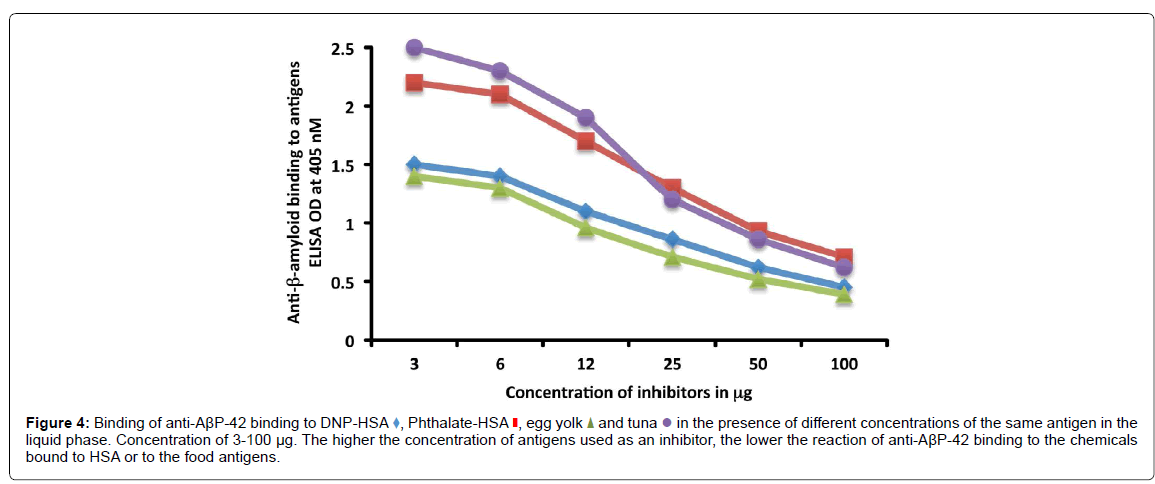
Figure 4:Binding of anti-AßP-42 binding to DNP-HSA , Phthalate-HSA, egg yolk and tuna in the presence of different concentrations of the same antigen in the liquid phase. Concentration of 3-100 µg. The higher the concentration of antigens used as an inhibitor, the lower the reaction of anti-AßP-42 binding to the chemicals bound to HSA or to the food antigens.
For the proper quantification and demonstration of anti-AβP-42 antibody binding to AβP-42 peptide, known concentrations of AβP-42 peptide were used in Sandwich ELISA to generate a standard curve, showing an increase in proportion to the concentration of AβP-42, with a resulting correlation coefficient of 0.998 (Figure 5A). Furthermore, AβP-42, tau protein, α+β casein, and GAD-65 were diluted in serum diluent buffer to obtain final concentrations of 31.25, 62.5, 125, 500 and 1000 pg/mL which were then measured in the assay against the standard curve (Figure 5B). The obtained measurements were compared to the known concentrations as follows: 1065 pg/mL obtained vs 1000 expected with a variation of 7%; 487 pg/mL obtained vs 500 expected with a variation of 3%; 264 pg/mL obtained vs 250 expected with a variation of 6%; 122 pg/mL obtained vs 125 expected with a variation of 2%; 63 pg/mL obtained vs 62.5 expected with a variation of 1%; and 36 pg/mL obtained vs 31.25 expected with a variation of 15%.
We also used the same progressive concentrations of tau protein, α+β casein, and GAD- 65 in the same assay to examine the possible binding of AβP-42 antibodies to this protein and these peptides. We found that while AβP-42 antibodies did not bind to GAD-65, they did bind to higher concentrations of tau protein and α+β casein (Figure 5).
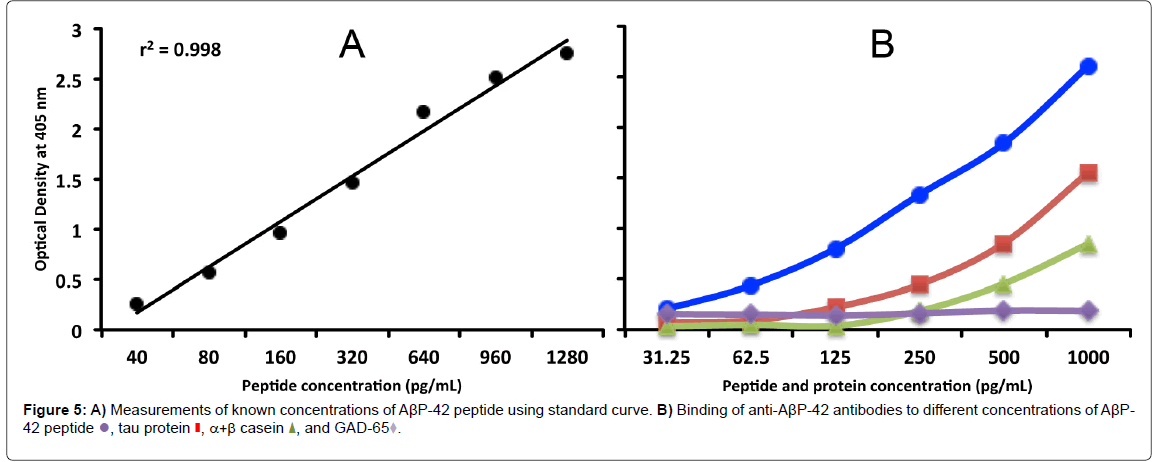
Figure 5:A) Measurements of known concentrations of AßP-42 peptide using standard curve. B) Binding of anti-AßP-42 antibodies to different concentrations of AßP- 42 peptide , tau protein , a+ß casein, and GAD-65.
Statistical Analysis
Statistical analysis using Microsoft Excel’s t-test function was performed. Comparison of the mean ± 3SD of the OD of all wells used as controls to the mean of the AβP-42 antibody reactivity against chemicals bound to HSA and all 208 tested food antigens was performed, and the p values are shown in the captions for Figures 1 and 2.
Discussion
In one of our previous review articles we dealt extensively with the potential link between autoimmune diseases and environmental triggers such as infectious diseases, toxic chemicals, and food proteins [59]. More specifically, our very recent article [60] focused on infectious agents involved in AD and their reaction with AβP antibody. This current study looks at the other two environmental triggers, and attempts to determine whether there is any connection between AD and the homology between some food proteins and AβP- 42 autoantibodies, as well as whether there is any immunoreactivity between these antibodies and toxic chemicals that are bound to human tissue proteins. We found that monoclonal anti- AβP-42 reacts from moderately to strongly with several chemicals bound to HSA, but not to many other chemicals bound to HSA, nor to HSA alone. This antibody also reacted with 19 out of the 208 food antigens used in the assay. Autoantibodies against β-amyloid protein and peptides are commonly detected in AD and even in some non-demented members of the ageing population [47]. The exact source of these anti-AβP antibodies is not clear, but they could be derived from immune response to aggregated forms of β-amyloid, from protein misfolding, or from antibodies to completely different antigens that cross-react with AβP-42. Aluminum, phthalate and dinitrophenyl are three chemicals bound to HSA that reacted significantly with anti-AβP-42. Because aluminum compounds are added to so many commercially-prepared products, from colored candies, cheese, coffee whiteners, to even drinking water, aluminum uptake into the bloodstream begins in utero and continues throughout life [19-24]. Although most absorbed aluminum is excreted, some of it manages to bind to different tissue proteins, particularly in the intestinal mucosa and in the brain [61-65]. Moreover, aluminum accumulation in the brain affects the memory and cognition, alters synaptic activity, activates microglia, and promotes β-amyloid and neurofilament aggregation, all of which are hallmarks of neurodegenerative disorders [66]. Aluminum is one of the factors that accelerate AβP-42 monomer aggregation by cross-linking anionic amino acids contained in the AβP-42 sequence to form AβP-42 aggregates [67]. This was reviewed very elegantly by Kawahara [22]. This may be one explanation as to why high levels of antibodies to AβP-42 and other amyloid proteins are detected in patients with AD [45,46]. Similar mechanisms of action may be applied to the aluminum binding to human albumin, where the aluminum may affect the functional properties of albumin or other proteins, leading to the formation first of dipoles and then of clusters that may mimic AβP-42 oligomerization or protein misfolding similarities [68]. This explanation is supported by the findings that numerous age-related disorders are now recognized to be related to the accumulation of different misfolded proteins that result in the production of autoantibodies called anti-oligomer antibodies [69].
Additional studies are needed to determine whether or not the addition of aluminum to albumin indeed results in protein misfolding, and if its injection into animal models results in the production of antioligomer antibodies that will react with AβP-42 and other associated misfolded proteins and peptides.
A similar interpretation may be applied to DNP-HSA or the plasticizer diethylphthalate-HSA, the neo-antigens to which anti- AβP-42 has reacted strongly (Figure 1). Using molecular modeling, the binding of phthalate plasticizers to HSA was examined in vitro. Fluorescence quenching data revealed that interaction of phthalate with HSA resulted in alterations in the conformational and secondary structures of HSA. Thermodynamic analysis also showed that hydrophobic sources were the main interaction for phthalates with HSA-protein [54].
Mercury is another chemical which extensive epidemiological and demographical studies have reported to have a strong association with AD [25]. In fact, according to one review article, some autopsy studies found increased levels of mercury in the brain tissues of AD patients but not in the blood, urine, hair or cerebrospinal fluid [25]. Furthermore, in in vitro and animal studies it was demonstrated that mercury causes tau protein phosphorylation, and the increased formation of amyloid-β protein [25]. Similar structural changes to HSA molecules due to mercury binding could be a plausible explanation for the detection of immune reactivity between AβP-42 antibody and mercury-HSA reported in our study. This is supported by the findings that, the same antibody did not react with HSA alone, formaldehyde- HSA, aflatoxin- HSA, and many additional chemicals bound to HSA that were used in our study. Further studies are needed to examine the differences between these two groups of chemicals, one group to which the AβP-42 antibody reacted strongly, and the second group to which the AβP-42 antibody did not (Figure 1).
However, it is much easier to explain the reaction of anti-AβP-42 antibody to PDI, an enzyme that is known to be the target for endocrine disruptors such as BPA and phthalates [34,35]. This binding of anti- AβP-42 with PDI may result in the loss of the latter’s functionality, and contribute to the α-synuclein aggregation and the production of the antibodies that are detected in AD [70] and Parkinson’s disease [71]. In relation to food association with AD, almost all the published articles are about proper diets that could cut the risk of AD, such as the MIND diet, which stands for “Mediterranean-dash Intervention for Neurodegenerative Delay [72].” Since, in our recent studies [58,73], we demonstrated that polyclonal and monoclonal antibodies made against the thyroid hormones T3 and T4 or against islet cell antigen reacted strongly with more than 30 different food proteins, we extended this approach to proteins and peptides that are target sites of AD. Using the binding of anti-AβP-42 antibody to AβP-42 at an OD of 3.8 as 100%, we found that 19 food antigens out of 208 different food extracts demonstrated moderate to strong immune reactivity with ODs ranging from egg yolk (1.55) to wheat globulin (3.2). This is equivalent to 38% and 84% of the immune reactivity of anti-AβP-42 with pure AβP-42 peptide. We were not surprised by these findings that certain foods may cross-react with neuronal cell antigens. In our own laboratory and others it has been shown that wheat peptide shares a significant homology with cerebellar and synapsin [74-77], with casein and butyrophilin, with MBP and myelin oligodendrocyte glycoprotein [77,78], and that plant aquaporins from tomato, corn, soy and spinach share up to 70% homology with human aquaporin or water channel proteins that are expressed in astrocytic foot processes [79,80]. It is therefore believed that these foods may be associated with gluten ataxia [81], multiple sclerosis [80], and neuromyelitis optica [79]. Because the extract of canned tuna, but not raw tuna, was the most reactive with anti-AβP-42, we repeated the experiment three times, starting with the tuna in the can all the way to protein extraction and ELISA assay, performing all in quadruplicates. With a plus or minus of 20% in ELISA ODs, all three canned tuna extract preparations gave strong reactions (a mean OD of 2.8 ± 0.36) with AβP-42 antibody. Then we extended the same experiment to raw tuna, since canned tuna goes through many modifications. We were very surprised to find out that the reaction of the AβP-42 antibody with raw tuna resulted in an OD of 0.80 ± 0.13. This reaction of canned tuna extract with anti-AβP-42 was an OD of 2.8 ± 0.27. This means that the mercury in the tuna and possibly the aluminum and plasticisizers used in the can may have changed the molecular structure of some tuna protein(s) so that the result is a strong reaction with the AβP-42-specific antibody. As was reviewed and shown by Kawahara [22], aluminum is an excellent cross-linker of proteins through three different metal-binding amino acids (arginine, thyrosine, and histidine).
Moreover, in our earlier research [82] we showed that certain foods after cooking and processing do become more antigenic, and so an individual may react to the cooked or modified food extract, but not to the raw version of the same food. Tuna proteins in the can are exposed to plasticizer, aluminum, and possibly other chemicals; we believe therefore that this heightened immune reactivity of AβP-42 antibody with cooked tuna is the result of the synergistic effect of mercury, aluminum, and plasticizer, compounded by the processes of cooking. This may have resulted in changes in the protein molecular structure of the canned tuna that made it more easily recognized by the AβP-42 antibody. Further study is needed to determine whether or not this crossreaction between AβP-42 antibody and chemicals bound to HSA, other possible proteins, and numerous foods shown in (Figure 2) does indeed play a role in amyloidogenesis. Stanyon and Viles in 2012 [83] observed a significant role for HSA in regulating amyloid-beta fibril growth in the brain interstitium. Although concentrations of Aβ found in blood plasma is high (0.1-0.5 mm), Aβ deposits are largely found in the brain. This is because HSA binds with 95% of Aβ in the blood and inhibits plaque formation in peripheral tissue. HSA also binds with endogenous fatty acids, especially hydroxynonenal, which can penetrate the BBB and then reach the brain, where, through the induction of oxidative stress, it may contribute to senile plaque formation, especially in ApoE4 individuals [84]. Furthermore, HSA is known for its ability to bind with additional molecules such as pharmaceuticals, aluminum and heavy metals, as shown in our study. Similar to fatty acids binding to HSA, the binding of toxic chemicals to HSA can result in BBB penetration and the deposition of these molecules’ antibodies and immune complexes into the amyloid plaque. This binding of aluminum, mercury or other haptenic molecules to HSA may compete with Aβ binding to HSA, resulting in a decrease in the concentration of active HSA on one hand, but the availability of a higher level of Aβ on the other hand. Based on this mechanism of action of HSA in the prevention of amyloid plaque formation, plasma exchange for increasing the level of HSA is used in clinical trials to reduce the levels of Aβ in the blood, hence reducing Aβ penetration into the brain, and inhibiting amyloid plaque formation [85,86] For now the results of our research provide a brief list of chemicals that can change human protein (HSA) so as to be recognized by the anti-AβP-42 antibody, as well as a menu of dietary proteins the antibodies of which may immunologically impact amyloid-β protein oligomerization; removal of these food items plus those shown by Carter [49] which share a homology with β-amyloid may be recommended at least in patients in the early stages of AD. Furthermore, as has been recommended in earlier studies [24-26,32], the most effective public health preventive measure for neurodegenerative disorders would be the elimination of mercury, aluminum, plasticizers and other toxic chemicals from human contact.
It is possible, based on the results of our study and the existing accumulated literature and knowledge, to hypothesize a mechanism that takes into account the effects of toxic chemicals such as aluminum, mercury, and phthalate plasticizers, and integrate then with the ability of some food antigen antibodies to cross-react with Aβ-42 in such a way so as to demonstrate a role in the amyloidogenesis that is characteristic of AD. To do this it is necessary to consider the blood-brain barrier (BBB).
The BBB is the regulated interface between the peripheral circulation and the central nervous system (CNS). The anatomical substrate of the BBB is the cerebral microvascular endothelium, which, together with astrocytes, pericytes, neurons, and the extracellular matrix, constitute a “neurovascular unit” that is essential for the health and function of the CNS. Tight junctions (TJ) between endothelial cells of the BBB restrict paracellular diffusion of water- soluble substances from blood to brain. The TJ is an intricate complex of transmembrane (junctional adhesion molecule-1, occludin, and claudins) and cytoplasmic (zonula occludens-1 and -2, cingulin, AF-6, and 7H6) proteins linked to the actin cytoskeleton [83,84,87,88]. It has been demonstrated that a number of different types of brain-reactive autoantibodies are not only numerous but ubiquitous in human serum in both healthy individuals and patients suffering from AD, although the levels are higher in subjects with AD [85,89]. While these autoantibodies can normally stay safely in the serum of a healthy person with an intact BBB, under certain conditions the BBB that normally protects the body by keeping out toxic environmental chemicals and unwanted molecules may fail or become permeable. The compromised BBB may then allow these brain-reactive autoantibodies, toxic chemicals, and cross-reactive food antibodies to gain access to the neurons within the brain tissue [85,86,89,90]. The brain-reactive autoantibodies may react with AβP-42 and other neural antigens,increasing intraneuronal amyloid-β deposition [87,91]. The toxic chemicals may have a direct effect on the brain, or the antibodies generated against them may bind to tissue proteins and form neo-antigens. Direct binding of the chemical to an Aβ-42 monomer results in the misfolding of the Aβ- 42 peptide, inducing amyloid fibril formation. Additionally, the food antibodies that have penetrated the BBB cross-react with Aβ-42, and may also bind to an Aβ-42 monomer, again resulting in in the misfolding of the Aβ-42 peptide, further contributing to amyloid fibril aggregation. In conjunction with the formation of immune complexes, the release of cytokines, activation of the complement cascade and other inflammatory factors, the amyloid fibril formation promotes the aggregation of senile plaques characteristic of Alzheimer’s disease. This synergistic mechanism combining all the above triggers and factors is shown in (Figure 6).
Figure 6:The synergistic effect of toxic chemicals and food-specific antibodies on amyloid plaque formation in Alzheimer’s disease. Toxic chemicals such as aluminum and mercury and food antibodies that cross-react with AßP-42 under certain conditions may penetrate the blood-brain barrier (BBB) and contribute to neurotoxic effects. A) BBB failure may result in the entry of toxic chemicals or of antibodies generated against chemicals which have bonded with tissue proteins (neo-antigens) into the nervous system. Direct binding of toxic chemicals to different proteins and peptides results in the formation of misfolded proteins, which in conjunction with the chemical antibody also binding to the misfolded proteins contributes to protein or peptide fibril formation. B) After crossing the BBB, AßP-42 cross-reactive food antibodies bind to an Aß peptide monomer and also induce amyloid fibril formation. The formation of immune complexes, release of cytokines, complement cascade activation, and other inflammatory factors further promote amyloid plaque aggregation, which contributes to AD neuropathology..
We believe that the results of our study show enough indications for immunoreactivity between AβP-42 autoantibodies, food proteins, and toxic chemicals in conjunction with a compromised BBB as having a role in the pathogenesis of Alzheimer’s disease. Effective prevention and early reversal requires knowing the status of each factor whether you have been exposed to toxic chemicals, pathogens, or cross-reactive foods that are involved in amyloidogenesis. We need to get to the cause of cognitive decline and fix any imbalances before the situation becomes irreversible. This means that in order to reverse cognitive decline in a neurodegenerative disease, it is necessary to identify and remove the triggering factors that are causing the brain’s defenses to misfire and produce a harmful instead of protective amyloid response.
The necessary processes can be summarized into these 3 steps:
1. Remove the triggers.
2. Remove the amyloid clusters.
3. Rebuild the synapses destroyed by the disease.
This idea of identifying the cause of the amyloid production, removing that, and then removing the amyloid (using monoclonal antibody) has not been tested, according to the book by Bredesen [88,92].
The linchpin of this idea, of course, is correctly identifying which factors are triggering the patient’s condition. Cross-reaction between AβP-42 and various infectious agents demonstrated in our earlier study [60] and with different food antigens shown in the present study indicate that circulating AβP-42 cross-reactive factors may interfere with the accurate measurements of AβP-42 levels in the blood. This, plus the presence of microbial-generated amyloids [88,93] in the blood of healthy subjects and patients with AD, may be a reason for reports of inconsistent results in the measurement of AβP-42 levels in serum [89,90,94,95]. It is also well known that the pathogenesis of typical Alzheimer’s disease is quite variable, depending on the age, genetic makeup, environmental exposure and the patient’s demographic. Due to these variables, it is possible that a certain percentage of the elderly may develop Alzheimer’s dementia even in the presence of a lower amyloid burden that is below the detectable threshold of amyloid-β plaque captured via PET imaging [96]. Due to the lower levels of amyloid-β peptide presented to cell-mediated immunity, these individuals may also exhibit lower levels of antibody to Aβ-42. Therefore, the presence of these and other antigens in the blood can interfere with accurate measurements of AβP-42 peptide and antibody levels.
That is why more research should be conducted to find reliable biomarkers for testing in the serum or cerebrospinal fluid of patients with Alzheimer’s disease.
Acknowledgment
The authors wish to thank Joel Bautista for the preparation of this manuscript for publication.
All financial and material support for this study was provided by the Corresponding Author.
References
- Coon KD, Myers AJ, Craig DW, Webster JA, Pearson JV, et al. (2007) A high-density whole-genome association study reveals that apoE is the major susceptibility gene for sporadic late-onset Alzheimer’s disease. J Clin Psychiatry 68: 613-618.
- Sadigh-Eteghad S, Talebi M, Farhoudi M (2012) Association of apolipoprotein E epsilon 4 allele with sporadic late onset Alzheimer`s disease. A meta-analysis. Neurosciences 17: 321-326.
- Liu CC, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy. Nat Rev Neurol 9: 106-118.
- Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, et al. (2016) Microbes and Alzheimer’s disease. J Alzheimers Dis 51: 979-984.
- Itzhaki RF (2014) Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front Aging Neurosci 6: 202.
- Zhan X, Stamova B, Jin L-W, DeCarli C, Phinney B, et al. (2016) Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 87: 2324-2332.
- Balin BJ, Hudson AP (2014) Etiology and pathogenesis of late-onset Alzheimer’s disease. Curr Allergy Asthma Rep 14: 417.
- Miklossy J (2015) Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front Aging Neurosci 7: 46.
- Alonso R, Pisa D, Marina AI, Morato E, Rábano A, et al. (2014) Fungal infection in patients with Alzheimer’s disease. J Alzheimer Dis 41: 301-311.
- Pisa D, Alonso R, Rabano A, Rabano A, Rodal I (2015) Different brain regions are infected with fungi in Alzheimer's disease. Sci Rep 5: 15015.
- Shoemark DK, Allen SJ (2015) The Microbiome and disease: Reviewing the links between the oral microbiome, aging, and Alzheimer’s disease. J Alzheimers Dis 43: 725-738.
- Kamer AR, Pirraglia E, Tsui W, Rusinek H, Vallabhajosula S, et al. (2015) Periodontal disease associates with higher brain amyloid load in normal elderly. Neurobiol Aging 36: 627-633.
- Ide M, Harris M, Stevens A , Sussams R, Hopkins V, et al. (2016) Periodontitis and cognitive decline in Alzheimer’s disease. Plos One 11: e0151081.
- Martande SS, Pradeep AR, Singh SP, Kumari M, Suke DK, et al. (2014) Periodontal health condition in patients with Alzheimer’s disease. Am J Alzheimer’s Dis Other Dement 29: 498-502.
- Syrjälä AM, Ylöstalo P, Ruoppi P, Komulainen K, Hartikainen S, et al. (2012) Dementia and oral health among subjects aged 75 years or older. Gerodontology 29: 36-42.
- Kim C, Lu G, Lee JS, Jung BC, Masuda-Suzukake M, et al. (2016) Exposure to bacterial endotoxin generates a distinct strain of a-synuclein fibril. Sci Rep 6: 30891.
- Asti A, Gioglio L (2014) Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J Alzheimers Dis 39: 169-179.
- Yegambaram M, Manivannan B, Beach TG, Halden RU (2015) Role of environmental contaminants in the etiology of Alzheimer’s disease: A review. Curr Alzheimer Res 12: 116-146.
- Campbell A (2002) The Potential role of aluminium in Alzheimer's disease. Nephrol Dial Transplant 2: 17-20.
- Exley C (2017) Aluminum should now be considered a primary etiological factor in Alzheimer’s disease. J Alz Dis Rep 1: 23-25.
- Kawahara M, Kato-Negishi M (2011) Link between aluminum and the pathogenesis of Alzheimer’s disease: The Integration of the aluminum and amyloid cascade hypotheses. Int J Alzheimers Dis 276393.
- Crapper DR, Krishnan SS, Dalton AJ (1973) Brain aluminum distribution in Alzheimer’s disease and experimental neurofibrillary degeneration. Science 180: 511-513.
- House E, Collingwood J, Khan A, Korchazkina O, Berthon G, et al. (2004) Aluminium, iron, zinc and copper influence the in vitro formation of amyloid fibrils of Aß42 in a manner which may have consequences for metal chelation therapy in Alzheimer’s disease. J Alzheimers Dis 6: 291-301.
- Haley E (2007) The Relationship of the toxic effects of mercury to exacerbation of the medical condition classified as Alzheimer's disease. Medical Veritas 4: 1510-1524.
- Mutter J, Curth A, Naumann J, Deth R, Walach H (2010) Does inorganic mercury play a role in Alzheimer’s disease? A systematic review and an integrated molecular mechanism. J Alzheimers Dis 22: 357-374.
- Mutter J, Naumann J, Sadaghiani C, Schneider R, Walach H (2004) Alzheimer disease: Mercury as pathogenetic factor and apolipoprotein E as a moderator. Neuro Endocrinol Lett 25: 331-339.
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, et al. (2005) The fetal basis of amyloidogenesis: Exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci 25: 823-829.
- Shih RA, Hu H, Weisskopf MG, Schwatrz BS (2007) Cumulative lead dose and cognitive function in adults: A review of studies that measured both blood lead and bone lead. Environ Health Perspect 115: 483-492.
- Suarez-Fernandez MB, Soldado AB, Sanz-Medel A, Vega JA, Novelli A, et al. (1999) Aluminum-induced degeneration of astrocytes occurs via apoptosis and results in neuronal death. Brain Res 835: 125-136.
- McGeer EG, McGeer PL (1998) The importance of inflammatory mechanisms in Alzheimer disease. Exp Gerontol 33: 371-378.
- Singh S, Li SS (2012) Epigenetic effects of environmental chemicals bisphenol A and phthalates. Int J Mol Sci 13: 10143-10153.
- Yeo M, Berglund K, Hanna M, Guo JU, Kittur J, et al. (2013) Bisphenol a delays the perinatal chloride shift in cortical neurons by epigenetic effects on the Kcc2 promoter. Proc Natl Acad Sci USA 110: 4315-4320.
- Cheng H, Wang L, Wang C (2010) Domain a’ of protein disulfide isomerase plays key role in inhibiting a-synuclein fibril formation. Cell Stress Chaperones 15: 415-421.
- Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, et al. (2006) S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441: 513-517.
- Borrell B (2010) Toxicology: The big test for bisphenol A. Nature 464: 1122-1124.
- Hajszan T, Leranth C (2010) Bisphenol A interferes with synaptic remodeling. Front Neuroendocrinol 31: 519-530.
- Braun JM, Kalkbrenner AE, Calafat AM, Yolton K, Ye X, et al. (2011) Impact of early-life bisphenol a exposure on behavior and executive function in children. Pediatrics 128: 873-882.
- Miodovnik A, Engel SM, Zhu C, Ye X, Soorya LV, et al. (2011) Endocrine disruptors and childhood social impairment. Neurotoxicol 32: 261-267.
- Téllez-Rojo MM, Cantoral A, Cantonwine DE, Schnaas L, Peterson K, et al. (2013) Prenatal urinary phthalate metabolites levels and neurodevelopment in children at two and three years of age. Sci Total Environ 461-462: 386-390.
- Xu H, Shao X, Zhang Z, Zou Y, Chen Y, et al. (2013) Effects of di-n-butyl phthalate and diethyl phthalate on acetylcholinesterase activity and neurotoxicity related gene expression in embryonic zebrafish. Bull Environ Contam Toxicol 91: 635-639.
- Sun W, Ban JB, Zhang N, Zu YK, Sun WX (2014) Perinatal exposure to di-(2-ethylhexyl)-phthalate leads to cognitive dysfunction and phosphor-tau level increase in aged rats. Environ Toxicol 29: 596-603.
- Vaiserman A (2014) Early-life exposure to endocrine disrupting chemicals and later-life health outcomes: An epigenetic bridge? Aging Dis 5: 419-429.
- Trudeau VL, Chiu S, Kennedy SW, Brooks RJ (2002) Octylphenol (OP) alters the expression of members of the amyloid protein family in the hypothalamus of the snapping turtle, chelydra serpentina serpentine. Environ Health Perspect 110: 269-275.
- Calderón-Garcidueñas L, Vojdani A, Blaurock-Busch E, Busch Y, Friedle A, et al. (2015) Air pollution and children: Neural and tight junction antibodies and combustion metals, the role of barrier breakdown, and brain immunity in neurodegeneration. J Alzheimer’s Dis 43: 1039-1058.
- Fox M, Knapp LA, Andrews PW, Fincher CL (2013) Hygiene and the world distribution of Alzheimer’s disease: Epidemiological evidence for a relationship between microbial environment and age-adjusted disease burden. Evol Med Public Health 173-186.
- Kellner A, Matschke J, Bernreuther C, Moch H, Ferrer I, et al. (2009) Autoantibodies against ß-amyloid are common in Alzheimer's disease and help control plaque burden. Ann Neurol 65: 24-31.
- Sohn JH, So JO, Hong HJ, Kim JW, Na DR, et al. (2009) Identification of autoantibody against beta-amyloid peptide in the serum of elderly. Front Biosci (Landmark Ed) 14: 3879-3883.
- Carter CJ (2010) Familial and late-onset Alzheimer's disease: Autoimmune disorders triggered by viral, microbial and allergen mimics of beta-amyloid and APP mutants? Nature Precedings.
- Carter C (2011) Alzheimer’s disease: APP, Gamma Secretase, APOE, CLU, CR1, PICALM, ABCA7, BIN1, CD2AP, CD33, EPHA1, and MS4A2, and their relationships with Herpes simplex, C. pneumoniae, other suspect pathogens, and the immune system. Int J Alzheimers Dis 501862.
- Carter C (2010) Alzheimer’s disease: A pathogenetic autoimmune disorder caused by Herpes simplex in a gene-dependent manner. Int J Alzheimers Dis 140539.
- Carter CJ (2017) Genetic, transcriptome, proteomic, and epidemiological evidence for blood-brain barrier disruption and polymicrobial brain invasion as determinant factors in Alzheimer’s disease. J Alzheimers Dis Rep 1: 125-157. Â
- Hatami A, Albay R, Monjazeb S, Milton S, Glabe C (2014) Monoclonal antibodies against Ab42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer disease brain. J Biol Chem 289: 32131-32143.
- Zhou XM, Lu WJ, Su L, Shan ZJ, Chen XG (2012) Binding of phthalate plasticizers to human serum albumin in vitro: A multispectroscopic approach and molecular modeling. J Agric Food Chem 60: 1135-1145.
- Lu F, Boutselis I, Borch RF, Hogenesch H (2013) Control of antigen-binding to aluminum adjuvants and the immune response with a novel phosphonate linker. Vaccine 31: 4362-4367.
- Yue Y, Zhang Y, Zhou L, Qin J, Chen X (2008) In vitro study on the binding of herbicide glyphosate to human serum albumin by optical spectroscopy and molecular modeling. J Photochem Photobiol B: Biology 90: 26-32.
- Â Vojdani A, Kharrazian D, Mukherjee PS (2015) Elevated levels of antibodies against xenobiotics in a subgroup of healthy subjects. J Applied Toxicol 35: 383-397.
- Kharrazian D, Herbert M, Vojdani A (2017) Immunological reactivity using monoclonal and polyclonal antibodies of autoimmune thyroid target sites with dietary proteins. J Thyroid Res.
- Vojdani A (2014) A potential link between environmental triggers and autoimmunity. Autoimmune Diseases.
- Vojdani A, Vojdani E, Saidara E, Kharrazian D (2018) Reaction of amyloid-ß peptide antibody with different infectious agents involved in Alzheimer’s disease. J Alzheimer’s Dis 63: 847-860.
- Walton RJ (2007) An aluminum-based rat model for Alzheimer’s disease exhibits oxidative damage, inhibition of PP2A activity, hyperphosphorylated tau, and granulovacuolar degeneration. J Inorg Biochem 101: 1275-1284.
- Pineton de Chambrun G, Body-Malapel M, Frey-Wagner I, Djouina M, Deknuydt F, et al. (2014) Aluminum enhances inflammation and decreases mucosal healing in experimental colitis in mice. Mucosal Immunol 7: 589-601.
- Walton RJ (2006) Aluminum in hippocampal neurons from humans with Alzheimer's disease. NeuroToxicol 27: 385-394.
- Anane R, Bonini M, Creppy EE (1997) Transplacental passage of aluminum from pregnant mice to fetus organs after maternal transcutaneous exposure. Hum Exp Toxicol 16: 501-504.
- Karlik SJ, Eichhorn GL (1989) Polynucleotide cross-linking by aluminum. J Inorgan Biochem 37: 259-269.
- Zhao Y, Hill JM, Bhattacharjee S, Percy ME, Pogue AID, et al. (2014) Aluminum-induced amyloidogenesis and impairment in the clearance of amyloid peptides from the central nervous system in Alzheimer’s disease. Front Neurol 5: 167.
- Saha A, Yakovlev VV (2010) Structural changes of human serum albumin in response to a low concentration of heavy ions. J Biophotonics 3: 670-677.
- Glabe G (2004) Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem Sci 29: 542-547.
- Nie CL, Wang XS, Liu Y, Perrett S, He RQ (2007) Amyloid-like aggregates of neuronal tau induced by formaldehyde promote apoptosis of neuronal cells. BMC Neuroscience 8: 9.
- Nagele E, Han M, DeMarshall C, Belinka B, Nagele R (2011) Diagnosis of Alzheimer’s disease based on disease-specific autoantibody profiles in human sera. Plos One 6: e23112.
- Han M, Nagele E, DeMarshall C, Acharya N, Nagele R (2012) Diagnosis of Parkinson’s disease based on disease-specific autoantibody profiles in human sera. Plos One7: e32383.
- Morris MC, Tangney CC, Wang Y, Sacks FM, Bennett DA, et al. (2015) MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement 11: 1007-1014.
- Kharrazian D, Herbert M, Vojdani A (2017) Detection of islet cell immune reactivity with low glycemic index foods-Is this a concern for type 1 diabetes? J Diabetes Res 4124967.
- Vojdani A, O’Bryan T, Green JA, McCandless J, Woeller KN, et al. (2004) Immune response to dietary proteins, gliadin and cerebellar peptides in children with autism. Nutr Neurosci 7: 151-161.
- Vojdani A (2015) Molecular mimicry as a mechanism for food immune reactivities and autoimmunity. Altern Ther Health Med 21: 34-45.
- Alaedini A, Okamoto H, Briani C, Wollenberg K, Shill HA, et al. (2007) Immune cross-reactivity in celiac disease: Anti-gliadin antibodies bind to neuronal synapsin I. J Immunol 178: 6590-6595.
- Vojdani A, Campbell AW, Anyanwu E, Kashanian A, Bock K, et al. (2002) Antibodies to neuron-specific antigens in children with autism: Possible cross-reaction with encephalitogenic proteins from milk, Chlamydia pneumoniae and Streptococcus group A. J Neuroimmunol 129: 168-177.
- Vojdani A, Kharrazian D, Mukherjee PS (2014) The prevalence of antibodies against wheat and milk proteins in blood donors and their contribution to neuroimmune reactivities. Nutrients 6: 15-36.
- Vaishnav RA, Liu R, Chapman J, Roberts AM, Ye H, et al. (2013) Aquaporin 4 molecular mimicry and implications for neuromyelitis optica. J Neuroimmunol 260: 92-98.
- Vojdani A, Mukherjee PS, Kharrazian D, Berookhim J (2015) Detection of antibodies against human and plant aquaporins in patients with multiple sclerosis. Autoimmune.
- Hadjivassiliou M, Sanders DS, Woodroofe N, Williamson C, Grünewald RA (2008) Gluten ataxia. Cerebellum 7: 494-498.
- Vojdani A (2009) Detection of IgE, IgG, IgA and IgM antibodies against raw and processed food antigens. Nutr Metab 6: 22.
- Stanyon HF, Viles JH (2012) Human serum albumin can regulate amyloid-ß peptide fiber growth in the brain interstitium: Implications for Alzheimer disease. J Biol Chem 287: 28163-28168.
- Bode DV, Stanyon HF, Hirani T, Baker MD, et al (2018) Serum albumin’s protective inhibition of amyloid-ß fiber formation is suppressed by cholesterol, fatty acids and warfarin. J Mol Biol 430: 919-934.
- Boada M, Ortiz P, Anaya F, Hernandez I, Munoz J, et al. (2009) Amyloid-targeted therapeutics in Alzheimer's disease: Use of human albumin in plasma exchange as a novel approach for Abeta mobilization. Drug News Perspect 22: 325-339.
- Mackic JB, Weiss MH, Miao W, Kirkman E, Ghiso J, et al. (1998) Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer’s amyloid /3 Peptide in aged squirrel monkey with cerebral amyloid angiopathy. J Neurochem 70: 210-215.
- Deane R, Zlokovic BV (2007) Role of the blood-brain barrier in the pathogenesis of Alzheimer's disease. Curr Alzheimer Res 4: 191-197.
- Â Hawkins BT, Davis TP (2005) The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev 57: 173-185.
- Levin EC, Acharya NK, Han M, Zavareh SB, Sedeyn JC, et al. (2010) Brain-reactive autoantibodies are nearly ubiquitous in human sera and may be linked to pathology in the context of blood-brain barrier breakdown. Brain Res 1345: 221-232.
- Clifford PM, Zarrabia S, Siu G, Kinsler KJ, Kosciuk MC, et al. (2007) Abeta peptides can enter the brain through a defective blood-brain barrier and bind selectively to neurons. Brain Research 1142: 223-236.
- Nagele RG, Clifford PM, Siu G, Levin EC, Acharya NK, et al. (2011) Brain-reactive autoantibodies prevalent in human sera increase intraneuronal amyloid-ß (1-42) deposition. J Alzheimers Dis 25: 605-622.
- Bredesen DE (2017) The end of Alzheimer’s: The first program to prevent and reverse cognitive decline, New York, Avery, Penguin Random House.
- Hill JM, Lukiw WJ (2015) Microbial-generated amyloids and Alzheimer’s disease (AD). Front Aging Neurosci 7: 9.
- Seppälä TT, Herukka SK, Hanninen T, Tervo S, Hallikainen M, et al. (2010) Plasma Abeta42 and Abeta40 as markers of cognitive change in follow-up: A prospective, longitudinal, population-based cohort study. J Neurol Neurosurg Psychiatry 81: 1123-1127.
- Mayeux R, Tang MX, Jacobs DM, Manly J, Bell K, et al. (1999) Plasma amyloid beta-peptide 1-42 and incipient Alzheimer’s disease. Ann Neurol 46: 412-416.
- Ossenkoppele R, Jansen WJ, Rabinovid GD, Knol DL, van der Flier WM, et al. (2015) Prevalence of amyloid PET positivity in dementia syndromes: A meta analysis. Jama 313: 1939-1949.
Citation: Vojdani A, Vojdani E (2018) Immunoreactivity of Anti-AβP-42 Specific Antibody with Toxic Chemicals and Food Antigens. J Alzheimers Dis Parkinsonism 8: 441. DOI: 10.4172/2161-0460.1000441
Copyright: © 2018 Vojdani A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 7244
- [From(publication date): 0-2018 - Nov 29, 2025]
- Breakdown by view type
- HTML page views: 6215
- PDF downloads: 1029