Spectroscopic and QSAR analysis on Antibiotic drug; 2-amino-4,6- dimethylpyrimidine using Quantum Computational Tools
Received: 01-Jan-2018 / Accepted Date: 23-Jan-2018 / Published Date: 27-Jan-2018 DOI: 10.4172/2329-9053.1000142
Abstract
The antibiotic activity of 2-amino-4,6-dimethylpyrimidine has been analyzed using molecular spectroscopy tools. The biological activity was interpreted and drug likeness was evaluated by calculating biological parameters. The activeness of the internal molecular parts was assessed by the assignment of fundamental modes of vibrations. The chromophores action for the inducement of the antibiotic activity of the compound was analyzed from the electronic excitation absorption peaks. The σ-bond, π-bond and δ-bond interaction lobes were identified and the exchange of energy between the orbitals was investigated from frontier molecular orbital profile. The asymmetrical charge distribution among different entities of the molecule for the perseverance of anti-tuberculosis mechanism was recognized. The NMBO interaction profile was evaluated by the NBO calculation adapted with Gaussian and the exchange of maximum energy transaction among various functional groups for the incentive of antibiotic were determined. The second order Polarizability of the compound emphasized the consistency of the antibiotic activity of the molecule. Thermodynamic activity of the molecule with respect to the temperature was stressed the decomposition rate and Gibbs free energy helped to determine the steadiness of the compound. The inhibition catalytic efficiency of the title molecule was fully tested by molecular docking study.
Keywords: 2-amino-4,6-dimethylpyrimidine; δ-bond interaction; NMBO; Antibiotic activity; Tuberculosis; Aermodynamic activity
Introduction
The pyrimidine belongs to the family of heterocyclic compounds nitrogen containing heterocycles are an important class of compounds in the medicinal chemistry occurs widely in living organisms (nucleic acids) and place unique position in heterocyclic and medicinal chemistry due to its useful biological activities and clinical applications[1,2]. Pyrimidine is biologically very imperative heterocyclic molecule which signified by most ubiquitous members of the diazine groups with Uracil and thymine being components of RNA and DNA and with cytosine. The pyrimidine compound is basically profuse in nature and is of great importance to human body since their structural subunits exist in many natural products; vitamins and antibiotics [3,4]. Hence, they have great attention in the design of antibiotic and biologically energetic compounds.
Usually, the Structural modification of antibiotic compounds enriched the antimicrobial resistance drugs extending the lifespan of drug agents [5]. The effect of substitutions making the structural modification and thus tri-substituted and tetra-substituted pyrimidine containing electron withdrawing group like amino group in pyrimidine were found to show more potent in vitro antimicrobial activity [6]. In addition to that, the further substitutions of methyl groups in different positions in the ring are improving the antifungal, anti-leishmanial and anti-inflammatory [7-9]. Thus, the methyl groups substituted in ortho and para positions in pyridine ring with amino group called 2-amino-4,6-dimethyl pyrimidine has enriched in vitro antibacterial and antitubercular activities and also antiviral agents such as the non-nucleoside reverse transcriptase inhibitors. It was also found that, the present molecule has well developed antimicrobial resistance which is currently available for chemotherapeutics usage [10].
There is high demand to analyze Pharmacodynamic activity and root cause of generation drug property on organic compositions for the immediate improvement of drug formulations in pharmacological field. However, after screening the current review 2-amino-4,6- dimethyl pyrimidine, there was no effort taken to analyze unknown properties and druglikeness with the help of molecular spectroscopy and computational tools. In this work, the biological parameters have been calculated and different analyses were made to focus the pharmaceutical activity of the present compound.
Material and Methods
Physical state
The compound has been taken in solid form which is pure and spectroscopic grade.
Recording profile
The FT-IR and FT-Raman spectra of the compound were recorded using a Bruker IFS 66V spectrometer and the instrument adopted with an FRA 106 Raman module equipped with aNd:YAG laser source operating at 1.064 μm line widths with 200 mW power [11].
The high resolution 1HNMR and 13CNMR spectra were recorded using 300 MHz and 75 MHz FT-NMR spectrometer [12].
The UV-Vis spectrum was recorded in the range of 100 nm to 800 nm, with the scanning interval of 0.2 nm, using the UV-1700 series instrument [13].
Computational profile
By fixing internal coordinate system, the present molecule was designed and by performing scanning process, the geometry was optimized. The high level hybrid calculations have been performed to calculate all the parameters which were used to carry over different analyses. The composite compound made by fusing ligand groups on base where the modified geometrical parameters, Mulliken charge displacement and the vibrational spectral properties were tabulated to study physical and chemical parameters. The entire quantum chemical computations were carried out by Gaussian 09 D. 01.version software in upgraded computer [13].
The computational calculations were performed using B3LYP and B3PW91 methods adopted with 6-31++G(d, p) and 6-311++G(d, p) basis sets. The energy profile of present compound related with electronic spectra, the NBO and HOMO-LUMO energies were calculated using time-dependent SCF method. similarly, the 1H and 13C NMR chemical shifts with respect to TMS were calculated by GIAO method using I-PCM model in combination with B3LYP/ 6-311++G(d, p). The Mullikan charge data of the compound was calculated and charge levels have been keenly observed for the elucidation of pharmaceutical activity of the compound. The dipole moment, linear Polarizability and the first order hyper-Polarizability in different coordinate system of the compound were computed using B3LYP method with the 6-311++G(d, p) basis set. The ECD and VCD spectra were simulated from available frequencies and the optical chirality was studied and the mechanism for masking the toxicity was interpreted.
Results And Discussion
Molecular deformation setup
The formation of covalent bonds among different atoms are usually affected the charge distribution among molecular site which leads intensive polarization of charges and making inductive effect. This effect causes certain degree of polarity in the bond which in term renders the bond length and angle much more liable to be compressed or elongated and thus the total geometry is altered by the intermolecular coulomb forces of attraction and repulsion. This ensures the existence of substitutional atoms and groups on aromatic base system and the effect of additions is pronounced by attainment of altered structure. Such asymmetrical proton and electron delocalization in atoms causing the stabilization of molecular structure which setup the chemical potential to generate drug activity.
Here, an amine group and couple of methyl groups were found to be identified on pyrimidine rings. The amine group was electron withdrawing element whereas the methyl group was identified as electron donor. The pyrimidine ring is already self-consistent electron containing aromatic frame and its consistency is usually disturbed by these types of substitutions. In this case, twelve heteronuclear bonds were appeared which is greater than homonuclear. Due to the substitutions, the bond lengths may be altered which would have positively and negatively charged with respect to the ligand attached with it.
The optimized structure of title molecule was showed in Figure 1 and the structural parameters have been presented in Table 1. The C-C bonds and C-N bonds in the main frame of pyrimidine rings are usually ranged between 1.375-1.383Å and between 1.329-1.344 Å respectively [14,15]. Here, the C-C bond lengths were observed to be 1.393Å and C-N bond lengths were appeared to be 1.340Å and 1.334Å respectively. The impact of substitution on ring was pronounced by the change of C-C and C-N bond lengths of 0.016Å and 0.011Å respectively. In the ring, due to the bond length change (C-N-1.340Å and C-C-1.393Å), the negative and positive inductive effects were observed in C-N and C-C bond lengths respectively.
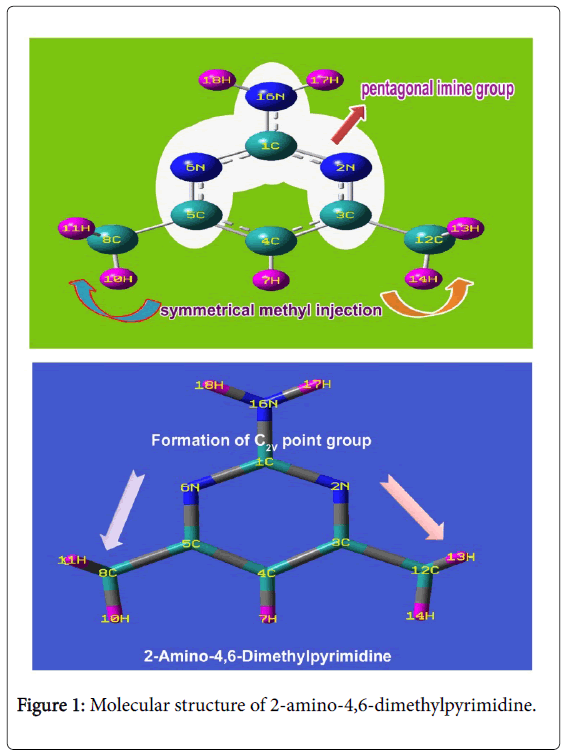
Figure 1: Molecular structure of 2-amino-4,6-dimethylpyrimidine.
| Geometrical Parameters | Methods | ||||
|---|---|---|---|---|---|
| HF | B3LYP | B3PW91 | |||
| 6-311++ | 6-31++ | 6-311++ | 6-31++ | 6-311++ | |
| G(d, p) | G(d, p) | G(d, p) | G(d, p) | G(d, p) | |
| Bond length(R) | |||||
| C1-N2 | 1.322 | 1.342 | 1.34 | 1.34 | 1.338 |
| C1-N6 | 1.322 | 1.342 | 1.34 | 1.34 | 1.338 |
| C1-N16 | 1.359 | 1.368 | 1.367 | 1.364 | 1.364 |
| N2-C3 | 1.318 | 1.336 | 1.334 | 1.334 | 1.331 |
| C3-C4 | 1.384 | 1.397 | 1.393 | 1.395 | 1.391 |
| C3-C12 | 1.5 | 1.504 | 1.502 | 1.499 | 1.497 |
| C4-C5 | 1.384 | 1.397 | 1.393 | 1.395 | 1.391 |
| C4-H7 | 1.069 | 1.082 | 1.079 | 1.083 | 1.08 |
| C5-N6 | 1.318 | 1.336 | 1.334 | 1.334 | 1.331 |
| C5-C8 | 1.5 | 1.504 | 1.502 | 1.499 | 1.497 |
| C8-H9 | 1.082 | 1.093 | 1.09 | 1.093 | 1.091 |
| C8-H10 | 1.08 | 1.091 | 1.088 | 1.091 | 1.088 |
| C8-H11 | 1.082 | 1.093 | 1.09 | 1.093 | 1.091 |
| C12-H13 | 1.082 | 1.093 | 1.09 | 1.093 | 1.091 |
| C12-H14 | 1.08 | 1.091 | 1.088 | 1.091 | 1.088 |
| C12-H15 | 1.082 | 1.093 | 1.09 | 1.093 | 1.091 |
| N16-H17 | 0.99 | 1.006 | 1.004 | 1.005 | 1.004 |
| N16-H18 | 0.99 | 1.006 | 1.004 | 1.005 | 1.004 |
| Bond angle(º) | |||||
| N2-C1-N6 | 126.464 | 126.673 | 126.566 | 126.883 | 126.784 |
| N2-C1-N16 | 116.762 | 116.656 | 116.707 | 116.549 | 116.598 |
| N6-C1-N16 | 116.759 | 116.653 | 116.709 | 116.55 | 116.6 |
| C1-N2-C3 | 116.712 | 116.472 | 116.525 | 116.312 | 116.364 |
| N2-C3-C4 | 121.604 | 121.368 | 121.314 | 121.475 | 121.413 |
| N2-C3-C12 | 116.465 | 116.584 | 116.649 | 116.526 | 116.613 |
| C4-C3-C12 | 121.93 | 122.047 | 122.035 | 121.998 | 121.972 |
| C3-C4-C5 | 116.895 | 117.637 | 117.747 | 117.535 | 117.653 |
| C3-C4-H7 | 121.551 | 121.18 | 121.125 | 121.231 | 121.172 |
| C5-C4-H7 | 121.551 | 121.18 | 121.125 | 121.231 | 121.172 |
| C4-C5-N6 | 121.603 | 121.368 | 121.315 | 121.475 | 121.413 |
| C4-C5-C8 | 121.928 | 122.045 | 122.035 | 122 | 121.972 |
| N6-C5-C8 | 116.467 | 116.585 | 116.649 | 116.524 | 116.613 |
| C1-N6-C5 | 116.713 | 116.472 | 116.523 | 116.31 | 116.362 |
| C5-C8-H9 | 109.554 | 109.966 | 109.98 | 109.913 | 109.947 |
| C5-C8-H10 | 111.746 | 111.768 | 111.76 | 111.862 | 111.811 |
| C5-C8-H11 | 109.59 | 110.005 | 110.014 | 109.964 | 109.979 |
| H9-C8-H10 | 109.145 | 108.935 | 108.931 | 108.94 | 108.959 |
| H9-C8-H11 | 107.562 | 107.029 | 107.042 | 106.984 | 107.004 |
| H10-C8-H11 | 109.145 | 109.008 | 108.986 | 109.045 | 109.009 |
| C3-C12-H13 | 109.59 | 110.004 | 110.013 | 109.965 | 109.981 |
| C3-C12-H14 | 111.747 | 111.77 | 111.76 | 111.859 | 111.811 |
| C3-C12-H15 | 109.554 | 109.968 | 109.981 | 109.913 | 109.945 |
| H13-C12-H14 | 109.144 | 109.007 | 108.982 | 109.04 | 109.012 |
| H13-C12-H15 | 107.562 | 107.028 | 107.044 | 106.987 | 107.002 |
| H14-C12-H15 | 109.146 | 108.936 | 108.932 | 108.943 | 108.959 |
| C1-N16-H17 | 116.281 | 117.027 | 116.755 | 116.892 | 116.608 |
| C1-N16-H18 | 116.28 | 117.026 | 116.759 | 116.896 | 116.611 |
| H17-N16-H18 | 117.231 | 118.231 | 117.915 | 118.238 | 117.942 |
| Dihedral angle(º) | |||||
| N6-C1-N2-C3 | 0.862 | 0.868 | 0.935 | 0.895 | 0.958 |
| N16-C1-N2-C3 | -177.71 | -177.61 | -177.51 | -177.59 | -177.5 |
| N2-C1-N6-C5 | -0.862 | -0.867 | -0.933 | -0.894 | -0.958 |
| N16-C1-N6-C5 | 177.713 | 177.611 | 177.507 | 177.591 | 177.495 |
| N2-C1-N16-H17 | -18.446 | -16.229 | -17.029 | -16.425 | -17.245 |
| N2-C1-N16-H18 | -162.84 | -165.14 | -164.32 | -164.87 | -164.09 |
| N6-C1-N16-H17 | 162.836 | 165.135 | 164.372 | 164.927 | 164.139 |
| N6-C1-N16-H18 | 18.443 | 16.226 | 17.083 | 16.484 | 17.296 |
| C1-N2-C3-C4 | -0.191 | -0.179 | -0.197 | -0.18 | -0.193 |
| C1-N2-C3-C12 | 179.885 | 179.896 | 179.903 | 179.886 | 179.913 |
| N2-C3-C4-C5 | -0.376 | -0.406 | -0.432 | -0.424 | -0.456 |
| N2-C3-C4-H7 | 179.969 | -179.99 | -179.99 | -179.97 | -179.99 |
| C12-C3-C4-C5 | 179.542 | 179.513 | 179.461 | 179.504 | 179.431 |
| C12-C3-C4-H7 | -0.111 | -0.066 | -0.099 | -0.043 | -0.105 |
| N2-C3-C12-H13 | 58.592 | 57.713 | 57.991 | 57.336 | 57.845 |
| N2-C3-C12-H14 | 179.706 | 178.952 | 179.199 | 178.65 | 179.102 |
| N2-C3-C12-H15 | -59.202 | -59.922 | -59.678 | -60.193 | -59.732 |
| C4-C3-C12-H13 | -121.33 | -122.21 | -121.91 | -122.6 | -122.05 |
| C4-C3-C12-H14 | -0.217 | -0.97 | -0.698 | -1.281 | -0.79 |
| C4-C3-C12-H15 | 120.874 | 120.154 | 120.423 | 119.874 | 120.375 |
| C3-C4-C5-N6 | 0.376 | 0.407 | 0.434 | 0.425 | 0.457 |
| C3-C4-C5-C8 | -179.54 | -179.52 | -179.46 | -179.51 | -179.43 |
| H7-C4-C5-N6 | -179.97 | 179.986 | 179.995 | 179.972 | 179.994 |
| H7-C4-C5-C8 | 0.109 | 0.064 | 0.096 | 0.037 | 0.107 |
| C4-C5-N6-C1 | 0.191 | 0.178 | 0.194 | 0.179 | 0.191 |
| C8-C5-N6-C1 | -179.88 | -179.9 | -179.9 | -179.88 | -179.92 |
| C4-C5-C8-H9 | -120.86 | -120.12 | -120.31 | -119.83 | -120.41 |
| C4-C5-C8-H10 | 0.232 | 1.005 | 0.805 | 1.325 | 0.76 |
| C4-C5-C8-H11 | 121.346 | 122.245 | 122.018 | 122.647 | 122.013 |
| N6-C5-C8-H9 | 59.215 | 59.957 | 59.782 | 60.234 | 59.701 |
| N6-C5-C8-H10 | -179.69 | -178.92 | -179.1 | -178.61 | -179.13 |
| N6-C5-C8-H11 | -58.578 | -57.68 | -57.884 | -57.291 | -57.878 |
Table 1: Optimized geometrical parameters for 2-Amino-4,6-Dimethyl pyrimidine computed at HF/DFT (B3LYP&B3PW91) with 6-31++G(d, p) & 6-311++G(d, p) basis sets.
Inter molecular bond lengths between methyl and amino groups were observed symmetrically which also make the molecule highly symmetry which also leads point group of symmetry to be C2V. This remarkable prototype of bond lengths may be associated with the small electronic resonance involving in the pyrimidine ring due to the ligand injection. The bottom moiety of ring frame of pyridine was found to be compressed much whereas top moiety was enlarged which was observed in the bond angle N2-C1-N6 (126.56°) and C3-C4-C5 (117.74°). The bond angle N2-C3-C4 and C4-C5-N6 were observed to be same as 121.31° which was due to the symmetrical association of methyl groups. The inductive and resonance effects among different entities ensured the substitutional effect on the generation of pharmaceutical property.
Mulliken charge assignment
The Mulliken charge display was presented in the Figure 2. Visualizing spatial distribution of electron density of a molecule in three dimensions is usually valuable and explaining forming of bond order. The Mulliken charge distribution profile of the compound explicit the molecular charge assignment with respect to the atomic charge enforcement due to intramolecular interaction of in phase and out of phase orbitals. This charge contour usually used to illustrate the chemical reaction forces which causing dynamic activity for generating the desired molecular chemical properties. The nucleophilic and electrophilic profile over the complex molecule displayed charge separation that showed driving chemical potential to induce antibiotic activity.
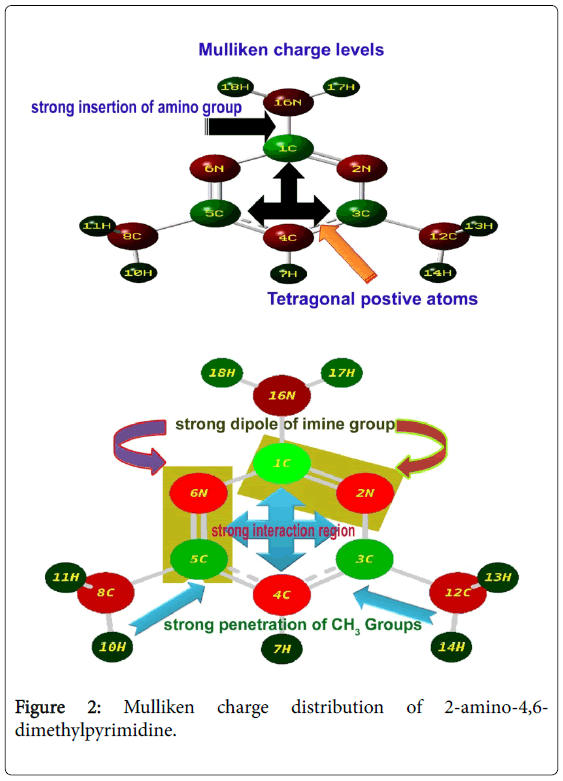
Figure 2: Mulliken charge distribution of 2-amino-4,6- dimethylpyrimidine.
The Mulliken partial charge distribution is always depends on interaction energy existed among atoms of molecular complex in which an electrostatic component (ionic), a Pauli repulsion component (steric) and an orbital relaxation component (covalent) are very significant to describe the cause of orientation of drug property. Here, four steric components were found on ring whereas all other bonds were identified as electrostatic components. Apart from that, there were some relaxation components were appeared. These arrangements of molecular interaction components ensured that, the strong insertion of amino groups. The electrostatic components emphasized the strong interactive imine groups in the ring which explicit the inducement of antibiotic property. The tetragonal charge depletion profile of the ring explored the driving potential for the sufficient inducement of antibiotic activity. The addition of amino group always enhanced the antibiotic activity in the molecular complex [16]. In this case, the strong injection of amino group emphasized the stability of antibiotic character. The addition of methyl groups also enhanced the charge depletion in the ring and observed to be control the acidity in the molecular complex which results sterilized antimicrobial resistance.
Molecular and biological property
The calculated Lipinski’s parameters and drug-likeness molecular properties of title compound using HyperChem 8.0.6 software were presented in Table 2. The well-constructed topographical polar surface area & lipophilicity profile drawing of title compound were showed in Figure 3. Lipinski rule of five also known as Pfizer's rule of five is basically used to determine whether the chemical compound with a certain pharmacological or biological activity is able to have orally active drug [17,18]. During the construction of drug, in order to improve the affinity and selectivity of drug, lipophilicity, non-rotatable bonds, ligand efficiency and molecular weight are increased; it is difficult process to maintain drug-likeness (RO5-fulfilment) throughout hit and lead optimization.
| Parameters | Values |
|---|---|
| Hydrogen bond donor count | 1 |
| Hydrogen bond acceptor count | 3 |
| Rotatable bond count | 0 |
| Topological Polar Surface Area | 168.4 |
| Mono isotopic Mass | 123.08 g/mol |
| Exact Mass | 123.08 g/mol |
| Heavy Atom Count | 9 |
| Covalently-Bonded Unit Count | 1 |
| Log P | 0.68 |
| TPSA | 51.81 |
| n atoms | 9 |
| Molecular Weight | 123.16 |
| nON | 3 |
| nOHNH | 2 |
| n violations | 0 |
| nrotb | 0 |
| Volume | |
| GPCR ligand | 2.08 |
| Ion channel modulator | 2.08 |
| Kinase inhibitor | 2.11 |
| Nuclear receptor ligand | 2.83 |
| Protase inhibitor | 2.6 |
| Enzyme inhibitor | 1.53 |
| Ligand efficiency | |
| Lipophilicity efficiency |
Table 2: Molecular properties and Bioactivity of 2-Amino-4,6- Dimethylpyrimidine.
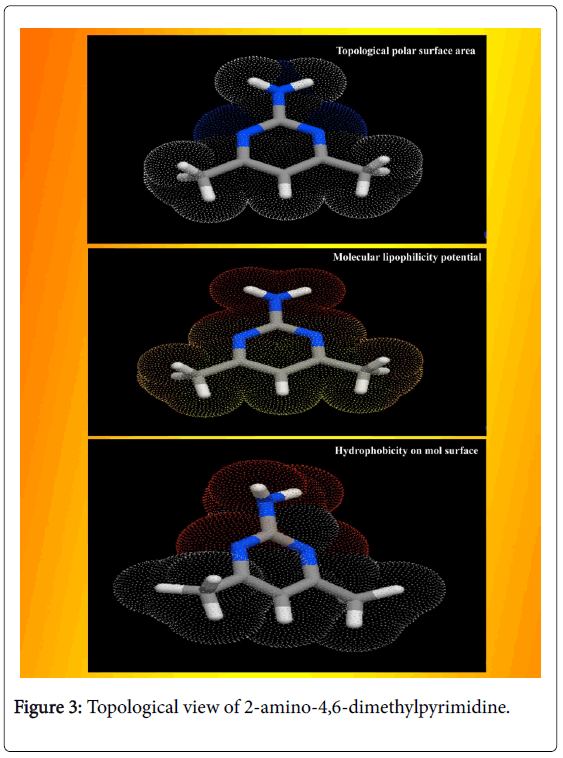
Figure 3: Topological view of 2-amino-4,6-dimethylpyrimidine.
If the aromatic drug complex is to have active oral, the rule of five (RO5) should have been satisfied. For that, the molecular weight should be less than 500 g/mol, the partition coefficient in octanolwater, Log P is better to less than 5, the Hydrogen bond acceptor is normal to less than 10 and Hydrogen bond Donor is enough to less than 5 [19]. In the present drug case, the Molecular Weight was found to be 123.16, the Log p was 0.68, the HBA count was observed to be 1 and the HBD value was found to be 1. The Lipinski rule of five (RO5) is most significant for the improved bioavailability; usually, only 20% of oral drugs disobey at least one of the criterion and 10% not be successful in two or three [20]. In this case, these observed values of such parameters describe that, the present molecule obeyed the Lipinski rule of five and pharmacologically active lead structure. Generally, if the molecule having the rotatable bonds ≤ 10 and total polar surface area ≤ 140 A2, the compound is able to possess good membrane permeability and oral bio availability for rule describes molecular properties important for a drug's pharmacokinetics [21]. Here, the RB and TPSA were calculated to be 0 and 51.81 A2 respectively. Due to the lesser values of such parameters, the present chemical compound can be passed in aqueous blood and break through the lipid-based cell membrane to arrive inside of a cell.
The heavy atom count of composition and Covalently-Bonded Count of present complex was determined to be 9 and 1 respectively. The CB count always maintained the covalent flavour of the chemical compound which was ensure the covalent character of the compound and the presence of heavy atom was N which consistently made imine combinations in ring which stressed the antibiotic activity. The secured score of drug likeness was evaluated to be -0.22 to -0.98 for the present molecular complex. Even though such the parameter range was identified to be negative, it was limited in adequate region which revealed that, this molecular complex will be a new drug candidate with adequate permeability.
The GPCR is G protein-coupled receptors (GPCRs) which is called 7TM receptors and G protein-linked receptors (GPLR) which is used to detect the ligand molecules outside the cell and activate internal signal transduction roots and it was found to be 2.08 for the present case. The observed value of GPCR showed the good signal transduction taking place due to the presence of ligand. The Ion channel modulator value was determined to be 2.08 for the title molecule which is comparatively adequate for pore-forming membrane proteins and is able to allow ions to pass through the channel pore. The kinase inhibitor is enzyme inhibitor which blocks the action of one or more protein kinases and its value was found to be 2.11. The observed value exposed its ability to modulate its function of protein kinases. The nuclear receptors are multifunctional protein that play important role in both embryonic development and adult homeostasis, for this case, it was found to be 2.83. This was relatively high and the present ligand can transduce signals of their cognate ligands. The Protease inhibitor is an antiviral capacity of the aromatic drug complex. The value was identified to be 2.60 and it was ensured that, the present compound has intensive antiviral activity.
Vibrational profile
The spectral sequence observed in IR and Raman is normally formulated for the identification of vibrational region of homo and hetero nuclear bonds composing aromatic structure. Usually, every molecular structure of aromatic compound is constructed by the amalgamation of base and substitutional groups and it is tailored in particular sequential form for obtaining desired chemical properties. The preferred medical property is achieved by adopting different atomic entities at internal coordinate system. By knowing the vibrational region, the presence of molecular bonds can be identified and the activeness of molecular entities showed the rate of operating mechanism originating to produce the particular drug property. For this interpretation, the FT-IR and FT-Raman frequency blueprint should be observed clearly. In this case, the spectra for the title compound was appeared to be fine and fundamental frequency pattern recognized in characteristic region of the each and individual bonds of molecule.
According to the mutual exclusion principle, the fundamental, group and substitutional frequencies of 2-Amino-4,6-Dimethyl pyrimidine were recorded and the assignments were made according to expected region with maximum accuracy. Due to the symmetrical substitutions of methyl groups on pyrimidine ring with axial injection of amino group, the present molecule belongs to C2V point group of symmetry. So, the fundamental modes of vibrations are possible to split up in to A1, B2, A2 and B1 vibrations and summed up in to 48 vibrations.
Usually, the A1 and B2 irreducible representations are assigned to stretching, ring deformation and in plane bending vibrations whereas the A2 and B1 allocated to ring, torsion and out of plane bending vibrations [22]. In order to study the dynamic activity of the bonds and bond angles, the FT-IR and FT-Raman frequencies were assigned carefully and the theoretical values were at higher level theories with appropriate basis sets which were presented in Table 3. The theoretical and observed vibrational pattern of FT-IR and FT-Raman spectra were illustrated in the Figures 4 and 5 respectively.
| Symmetry Species | Observed Frequency(cm-1) | Methods | Vibrational Assignments | |||||
|---|---|---|---|---|---|---|---|---|
| C2V | HF | B3LYP | B3PW91 | |||||
| FT-IR | FT-Raman | 6-311++G(d, p) | 6-31++G(d, p) | 6-311++G(d, p) | 6-31++ G(d, p) | 6-311++G(d, p) | ||
| A1 | 3380s | - | 3575 | 3571 | 3599 | 3591 | 3573 | (N-H) υ |
| A1 | 3360s | 3360m | 3464 | 3445 | 3445 | 3462 | 3453 | (N-H) υ |
| A1 | 3050s | - | 3046 | 3063 | 3058 | 3038 | 3057 | (C-H) υ |
| A1 | 2940s | - | 2949 | 2940 | 2948 | 2945 | 2945 | (C-H) υ |
| A1 | - | 2930s | 2949 | 2939 | 2948 | 2944 | 2944 | (C-H) υ |
| A1 | - | 2925s | 2933 | 2930 | 2924 | 2920 | 2920 | (C-H) υ |
| A1 | 2920s | - | 2933 | 2930 | 2924 | 2920 | 2920 | (C-H) υ |
| A1 | 2880s | - | 2882 | 2872 | 2875 | 2883 | 2877 | (C-H) υ |
| A1 | 2875s | - | 2882 | 2872 | 2874 | 2883 | 2877 | (C-H) υ |
| A1 | 1610s | - | 1613 | 1609 | 1611 | 1615 | 1610 | (N-H) δ |
| A1 | - | 1600s | 1602 | 1593 | 1600 | 1605 | 1600 | (N-H) δ |
| A1 | 1580vs | - | 1585 | 1582 | 1576 | 1572 | 1584 | (C=N) υ |
| B2 | 1475vs | - | 1479 | 1474 | 1477 | 1476 | 1475 | (C=N) υ |
| B2 | - | 1460m | 1456 | 1467 | 1458 | 1462 | 1464 | (C=C) υ |
| A1 | - | 1450m | 1448 | 1453 | 1446 | 1449 | 1451 | (C-N) υ |
| A1 | - | 1445m | 1446 | 1452 | 1445 | 1447 | 1448 | (C-N) υ |
| A1 | - | 1440m | 1446 | 1435 | 1440 | 1443 | 1432 | (C-N) υ |
| B2 | 1415w | - | 1411 | 1413 | 1423 | 1416 | 1417 | (C-C) υ |
| B2 | - | 1395m | 1391 | 1397 | 1393 | 1399 | 1398 | (C-C) υ |
| B2 | 1380vs | - | 1377 | 1384 | 1377 | 1371 | 1376 | (C-C) υ |
| B2 | 1325s | - | 1329 | 1321 | 1319 | 1319 | 1321 | (CH3)α |
| B2 | - | 1190w | 1192 | 1190 | 1193 | 1190 | 1190 | (CH3)α |
| A1 | - | 1150w | 1147 | 1155 | 1145 | 1150 | 1150 | (C-H) δ |
| A1 | - | 1080w | 1078 | 1079 | 1082 | 1081 | 1083 | (C-H) δ |
| A1 | 1060w | - | 1060 | 1063 | 1060 | 1066 | 1064 | (C-H) δ |
| A1 | - | 1050vw | 1051 | 1050 | 1049 | 1046 | 1048 | (C-H) δ |
| A1 | 1040w | - | 1037 | 1036 | 1042 | 1039 | 1042 | (C-H) δ |
| A1 | 1000w | - | 996 | 1002 | 995 | 997 | 999 | (C-H) δ |
| A1 | 980w | - | 979 | 983 | 976 | 976 | 977 | (C-H) δ |
| B1 | - | 950m | 946 | 947 | 949 | 944 | 945 | (N-H) γ |
| B1 | - | 920m | 914 | 920 | 919 | 915 | 924 | (N-H) γ |
| B1 | 850w | - | 818 | 823 | 818 | 817 | 818 | (C-H) γ |
| B1 | 820s | - | 800 | 803 | 801 | 802 | 800 | (C-H) γ |
| B1 | 710m | - | 653 | 653 | 654 | 652 | 651 | (C-H) γ |
| B1 | - | 700vs | 620 | 618 | 620 | 622 | 621 | (C-H) γ |
| B1 | 680s | - | 565 | 564 | 563 | 566 | 564 | (C-H) γ |
| B1 | - | 640w | 537 | 541 | 538 | 538 | 538 | (C-H) γ |
| B1 | - | 610w | 524 | 526 | 527 | 524 | 525 | (C-H) γ |
| B2 | 540m | - | 501 | 501 | 502 | 501 | 501 | (C-N) δ |
| B2 | - | 480w | 460 | 458 | 461 | 462 | 460 | (C-C) δ |
| B2 | - | 420w | 423 | 415 | 440 | 420 | 451 | (CNC) δ |
| B2 | - | 300w | 301 | 300 | 300 | 301 | 301 | (CNC) δ |
| B2 | - | 275w | 274 | 275 | 275 | 275 | 274 | (CCC) δ |
| A2 | - | 220w | 221 | 243 | 236 | 242 | 238 | (CNC) γ |
| A2 | - | 200w | 200 | 219 | 213 | 221 | 217 | (CNC) γ |
| A2 | 170w | - | 170 | 169 | 170 | 189 | 187 | (CCC) γ |
| A2 | 75w | - | 75 | 68 | 67 | 64 | 63 | (C-N) γ |
| A2 | 70w | - | 70 | 66 | 66 | 63 | 62 | (C-C) γ |
Table 3: observed and calculated vibrational frequencies of HF and DFT (B3LYP&B3PW91) with 6-31++(d, p) & 6-311++G (d, p) level 2- Amino-4,6-Dimethylpyrimidine.
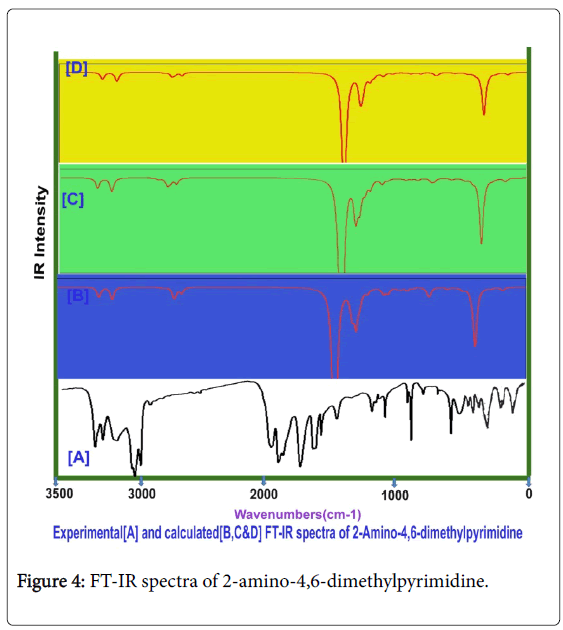
Figure 4: FT-IR spectra of 2-amino-4,6-dimethylpyrimidine.
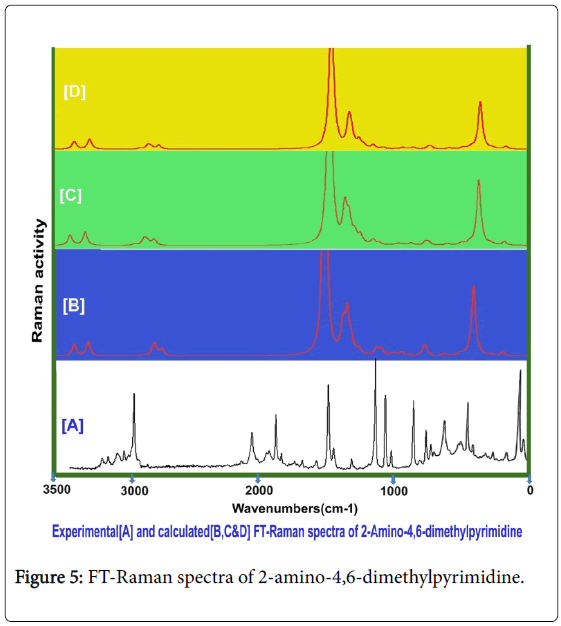
Figure 5: FT-Raman spectra of 2-amino-4,6-dimethylpyrimidine.
Aromatic and methyl C-H vibrations: As the present case is basically pyrimidine and has tri substituted system of compound, the ring possessed one unique C-H bond. Even though, this compound is heterocyclic, the aromatic C-H stretching, in plane and out of plane bending vibrations are usually observed in the region 3120-3010 cm-1, 1250-1000 cm1 and 950-720 cm1 respectively [23-25]. In this case, the C-H stretching, in plane and out of plane bending vibrational peaks were found with strong to weak intensity at 3050, 1150 and 820 cm1 respectively. All the observed bands of vibrations have been appeared at the middle portion of the expected region which explicited that, the entire vibrations neither affected nor influenced much.
The couple of methyl groups were injected strongly in symmetrical form in meta positions in pyrimidine ring. Due to the symmetrical substitutions, it was found that, there was no change in optimized parameters. By this effect, the corresponding vibrational pattern should be observed in sequential pattern. Accordingly, the related C-H stretching modes were identified with strong intensity at 2940, 2930, 2925, 2920, 2880 and 2875 cm1. Usually, these vibrational bands were observed with medium to weak intensity whereas in this case, all the bands were observed with strong intensity. Normally, these stretching bands are observed in the region of 2880-3000 cm1 [26,27]. The C-H in plane and out of plane bending modes for methyl group is appeared in the region 1250-950 cm1 and 950-680 cm1 respectively [28,29] for methyl added aromatic system. Hence, for this case, the in plane and out of plane bending vibrations were identified at 1080, 1060, 1050, 1040, 1000 and 980 cm1 and 820, 710, 700, 680, 640 and 610 cm-1 respectively. Except two vibrational modes, the entire bending peaks were observed within the expected region. From the methyl group vibrations, it was found that, the vibrational pattern emphasized the symmetrical presence of methyl groups in meta positions. From this worth points, it was concluded that, the energy of methyl groups were utilized much for control the acidity instead of the generation of drug property.
Core CC vibrations: For pyrimidine and its derivative compounds, the C=N and C=C stretching vibrational bands are conditionally located in the spectral region 1600-1500 cm1 [30,31]. For this case, the C=N is usually absorb the IR radiation in 1580-1520 cm-1 and C=C and C-C stretching bands are identified in the region 1480-1380 cm1. In this case, the C=N stretching modes were determined at 1580 and 1475 cm1, the C=C stretching mode is observed at 1460 cm1 and C-C stretching band has been found at 1415 cm-1. The imine group based core vibrations have been observed to be rather suppressed. Usually, the amino injection influences the core vibrations, but here, there was little effect of amino group observed. This bumbled state described that, the amino group energy was supported the energy of pyrimidine ring which leads the core vibrations to some extent. The CNC and CCC in plane bending modes were observed at 420 and 300 and 275 cm1 respectively. The out of plane bending modes were found at 220 and 200 cm-1and 175 cm1 respectively. These vibrational assignments have been agreed with above literatures.
Amino group vibrations: In present case, the primary amino group was substituted strongly and it has no adjacent effective ligand groups. The primary amine group vibrations always dominate in the spectral pattern of the parent compound. For aromatic amine group, one band of N-H stretching vibration is observed with medium intensity in the region 3520-3420 cm-1 which may be asymmetrical and another band is found in the region 3420-3340 cm1 [32,33] which may be symmetrical. Here, the stretching modes for N-H bond were found with strong intensity in IR and medium intensity in Raman at 3360 cm1 and 3360 cm1 respectively. According to the literatures, two stretching bands were identified to be symmetrical and observed very closely. The related in plane and out of plane bending vibrations with medium to strong intensity are usually determined in the region 1650-1580 cm-1 and 895-650 cm1 [34,35]. Accordingly, the in plane (Scissoring vibrations) bending peaks were determined at 1610 &1600 cm1 and 950 & 920 cm1 respectively. The in plane bending was observed well within the expected region whereas the out of plane bending modes were pushed up to the higher spectral region. This view was strongly represented the supremacy character of amino group and also control the sore vibrations. From this discussion, it was concluded that, the drug mechanism was operated by amino group.
C-N vibrations: In this case, the C-N bands were found at two places in the compound; one was from core ring and another was injection point of amino group. In the case of amine C-N, the stretching mode for the same is found in the region 1360-1280 cm1 [36] and for core C-N is observed usually in the region 1350-1250 cm1 [37]. In this present case, for core C-N, the stretching bands were observed at 1450 and 1445 cm-1 and for amine C-N, the oscillated vibration was found at 1440 cm-1. The entire C-N bond stretching modes were found to be elevated to well above the allowed region which showed its consistency in core ring and amine binding.
NMR examination
The NMR profile is not only used for deshielding effect of proton due to the strong couplings of neighbour atoms and also consumed for studying chemical reaction path mechanism to evaluate the accumulation of chemical potential causing drug root. The chemical shift of molecular carbon at different ambiance in the aromatic complex usually explicited the rate of activation of ligand with base compound and electron cloud dissociation index for blending of drug property through the interaction lobe.
The experimentally recorded and calculated isotropic chemical shift in gas as well as solvent phase was presented in the Table 4 and their simulated spectra were displayed in Figure 6. In the pyrimidine aromatic complex, the pyrimidine ring base was fused with amino group and methyl groups. Here, apart from the ring, there were two carbons found for methyl groups in which almost same chemical shift was observed. The carbon joined with amino group was occupied by three nitrogen atoms where in which very high chemical shift was found. The experimental chemical shift for ring carbons were determined in the region 110-168 ppm and the calculated values (119-185 ppm). But, for the methyl carbons, very low chemical shift was observed. The entire chemical shifts of the compound were observed to be well agreed with literature values [38].
| Atom position | Calculated shift in (ppm) B3LYP/6-311+G(2d, p) | Experimental shift (ppm) | ||
|---|---|---|---|---|
| Gas | Solvent phase | |||
| DMSO | CCl4 | |||
| 1C | 185.85 | 185.79 | 185.89 | 167 |
| 3C | 187.61 | 188.85 | 188.21 | 168 |
| 4C | 119.74 | 121.37 | 120.31 | 110 |
| 5C | 170.25 | 172.71 | 171.28 | 164 |
| 8C | 24 | 24.33 | 24.11 | 23 |
| 12C | 21.83 | 22.13 | 21.93 | 22 |
| 7H | 6.64 | 6.98 | 6.77 | 6.3 |
| 9H | 2.1 | 2.19 | 2.14 | 6.2 |
| 10H | 1.72 | 2 | 1.82 | 1.9 |
| 11H | 1.83 | 1.74 | 1.8 | - |
| 13H | 2.1 | 2.21 | 2.15 | 2.2 |
| 14H | 1.94 | 2.19 | 2.03 | 1.8 |
| 15H | 1.99 | 1.9 | 1.97 | - |
| 17H | 3.75 | 4.06 | 3.88 | 5.7 |
| 18H | 3.81 | 4.1 | 3.94 | 5.5 |
| 2N | 304.87 | 297.66 | 301.8 | - |
| 6N | 338.17 | 325.25 | 332.81 | - |
| 16N | 90.72 | 90.64 | 90.73 | - |
Table 4: Experimental and calculated chemical shifts (ppm) of 2-Amino-4,6-Dimethylpyrimidine.
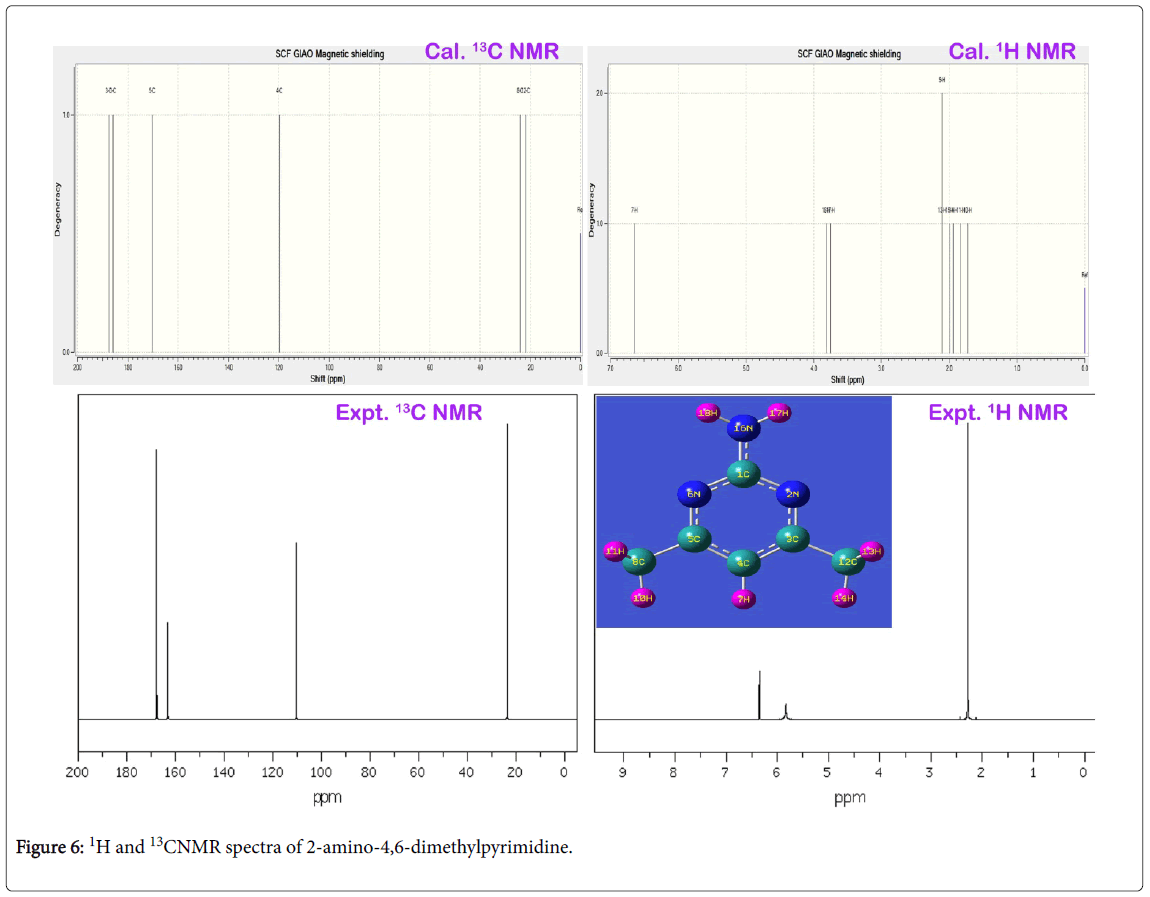
Figure 6: 1H and 13CNMR spectra of 2-amino-4,6-dimethylpyrimidine.
The carbon C1 was acting as bridge point for the coupling of amino group, the chemical shift of the same was 167 ppm. Since the electron cloud was partially shared by surrounding nitrogen atoms, the proton shielding of C1 was broken multiply and also its proton acted as main source for exchanging electron cloud between ring and amino group. This was emphasized by the vibrational analysis. The chemical shift of carbon C4 was found to be 110 ppm which was located bottom moiety of the ring. This moderate chemical shift was ensured the unidirectional flow of electron cloud with respect to C4. This view was ensured in the Mulliken charge profile. The chemical shift carbon C3 and C4 were recorded to be 168 and 164 ppm (cal. 187 & 170 ppm) respectively. These chemical shifts were rather differed from one another which were due to asymmetrical stagnation of electron cloud from the degenerate periodic orbitals. Apart from that, the electron cloud was sucked by C of methyl groups in order to control the passage of electronic potential. The lower chemical shift of around 23 ppm was observed for C8 and C12. This is mainly due to the C of the methyl groups were further shielded by intake electrons. The anisotropic chemical shift of N2 and N6 were 304 and 334 ppm which was very high when compared with N16 (cal. 90 ppm) of amino group. Since these two nitrogens were making imine group in the ring, they playing important role in the property construction. From the observed chemical shift, it was concluded that, the ring itself having drug source which was enhanced by the amine group. The addition of methyl groups were found to control and stabilize the drug consistency.
Frontier molecular interaction examination
The cascade combinations of atomic orbitals usually fabricate the molecular orbitals and the energy between Lewis base (HOMO) and Lewis acid (LUMO) always represent the chemical reactivity. HOMO could be simply donating electron density to form a bond (act as a Lewis base) or it could be oxidation whereas LUMO could be energetically receives the electron density or could be reduction. The HOMO and LUMO set up can interact with one another with respect to the degenerate coefficients. Normally, these interactive orbitals are making common spaced orbital lobes in which the electron density are dislocated with respect to chemical equilibrium forces. The electronic energy excited among molecular sites by which the orbitals between ligand and base compositions are constructed. These overlapped space orbitals accomplished the resultant chemical property which leads the compound to be active drug. The FMO constructed orbital view for present molecule was shown in the Figure 7 and the energy coefficients of orbitals were depicted in the Table 5.
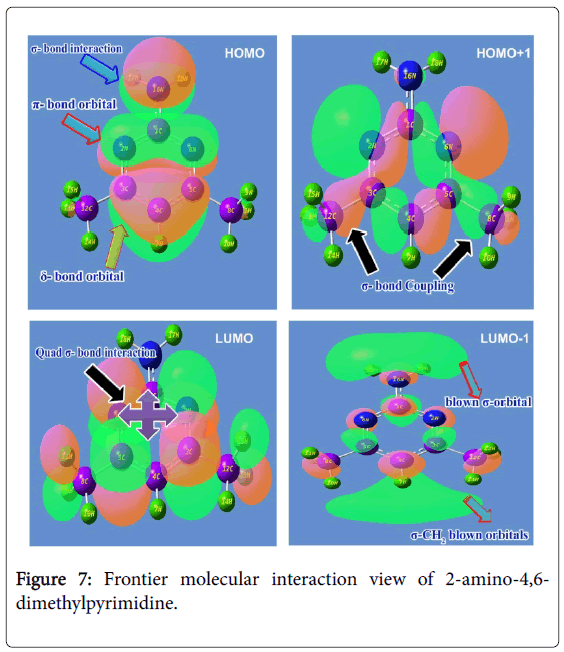
Figure 7: Frontier molecular interaction view of 2-amino-4,6- dimethylpyrimidine.
| Energy levels | B3LYP 6311++(d, p) Energy in ev | UV Gas Energy in ev |
|---|---|---|
| H+10 | 11.58034 | 11.82742 |
| H+9 | 11.31775 | 11.43558 |
| H+8 | 10.99285 | 11.14088 |
| H+7 | 10.98822 | 11.06605 |
| H+6 | 10.75829 | 10.85516 |
| H+5 | 10.31801 | 10.36236 |
| H+4 | 8.69975 | 8.8807 |
| H+3 | 8.42083 | 8.33212 |
| H+2 | 8.07225 | 7.94463 |
| H+1 | 6.83087 | 6.76311 |
| H | 6.34406 | 6.55875 |
| L | 1.00301 | 1.25743 |
| L-1 | 0.27728 | 0.51728 |
| L-2 | 0.15973 | 0.31891 |
| L-3 | 0.07537 | 0.04299 |
| L-4 | 0.17986 | 0.15211 |
| L-5 | 0.67021 | 0.683 |
| L-6 | 0.83021 | 0.72681 |
| L-7 | 0.87919 | 0.93198 |
| L-8 | 1.09961 | 1.06695 |
| L-9 | 1.26342 | 1.27349 |
| L-10 | 1.43839 | 1.48274 |
Table 5: Frontier molecular orbital’s with energy levels.
In this case, the Lewis base such as HOMO appeared over ring and amino group. The amino group was found to be constructed by σ- bonding interactions which were isolated from the ring. The in phase interaction was taking place among imine group of ring (N-C-N) which was determined to be constructed by π-bonding interactions. The δ-bonding interactions were found to be covered the semicircle (C-C-C) of lower moiety of pyrimidine ring. The electron density clouds that are able to offer the transitions were originated on the ring and NH2 species which could be contribute the chemical potential for establishing anti biotic potential. This chemical energy was found to be controlled by boundary of orbital lobes of reconstructed molecular orbitals.
The Lewis acid such as LUMO was established on C=N and C=C groups which was constructed by quad σ-bonding interactions. Apart from that, the σ-bonding interactions were observed over the H of methyl groups. The orbital space lobe of ring C was primarily connected with orbital lobe C of methyl groups by which the transitions are possible; this showed that, the involvement of chemical potential for stabilizing drug property. In second order HOMO, the positive and negative lobe interactions of C and N of the pyrimidine ring were blended with orbital lobe of C of methyl groups. This view was showed in different color. In second order LUMO, the Blown H orbital lobes were found top and bottom moiety which showed the blast orbital lobe.
UV-Visible absorption CT complex profile
The UV absorption band is consists of entire energy transitions among vibrational energy levels. Normally, the charge transfer complex (CT) is produced in the compound by substituting suitable ligand groups in the base molecule. The effect of substitutions results mechanism for inducing CT complex which is very significant to study the alternation of chemical property of base ring. The electronic absorption is observed for the chemical compound describes the effective ligands which acting as primary source for producing of desired chemical property. It is also used to find the rate at which the suitable ligand groups change of chemical property base molecule. The UV-Visible absorption band is appeared in the electronic spectra represents electronic excitation energy structured by the adoptive active ligands. The main objective of this study is to identify the CT ligands and rate of shift of electronic absorption peak. Consequently, it is very important to determine the rate of change of chemical property.
In this case, the electronic absorption band (three excitation levels) on the energy gap of 3.9, 4.7 and 4.8 eV at 311.94, 263.56 and 255.87 nm was identified respectively with the maximum oscillator strength 0.06. This band was found in gas phase which was represented by located by n→π* transitions and also this band were taking place in Quartz UV region of the spectrum. In solvent phase (DMSO), the band was found at 300, 256 and 255 nm with the energy gap of 4.1, 4.8 and 4.8 eV respectively. The strength of transitional oscillations was measured as 0.09 and is represented by n→π*. Similar to the gas phase, this band also located at UV-Visible region.
According to the theoretical aspect, this band was uniquely recognized as B and E band (German, radikalartig). In the case of present molecule, the observed electronic spectra showed three excited peaks at 248, 250 and 252 nm which were well coincide with calculated electronic excitations. The excited electronic and ECD spectra were displayed in the Figure 8. The electronic energy absorption data was presented in the Table 6.
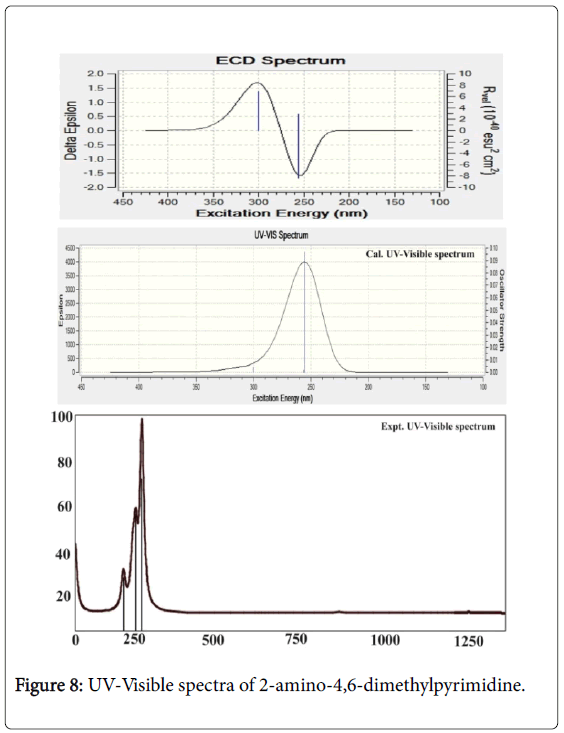
Figure 8: UV-Visible spectra of 2-amino-4,6-dimethylpyrimidine.
| Λ (nm) | E (eV) | ( f ) | Major contribution | Assignment | Region | Bands |
|---|---|---|---|---|---|---|
| Gas | B and E band (German, radikalartig) | |||||
| 311.9 | 3.974 | 0.003 | H→L (85%) | n→π* | Quartz UV | |
| 263.6 | 4.704 | 0.0013 | H→L-1 (92%) | |||
| 255.9 | 4.845 | 0.0648 | H+3→L (95%) | |||
| DMSO | ||||||
| 300.4 | 4.127 | 0.004 | H+1→L (85%) | n→π* | Quartz UV | |
| 256.4 | 4.835 | 0.0018 | H+2→L (92%) | |||
| 255.6 | 4.85 | 0.0969 | H+3→L (95%) | |||
| CCl4 | ||||||
| 307.5 | 4.033 | 0.0038 | H→L (85%) | n→π* | Quartz UV | |
| 260.7 | 4.756 | 0.0018 | H+2→L (92%) | |||
| 256.8 | 4.828 | 0.0939 | H+3→L (95%) | |||
Table 6: Theoretical electronic absorption spectra of 2-Amino-4,6-Dimethylpyrimidine (absorption symbols λ (nm), excitation energies E (eV) and oscillator strengths (f)) using TD-DFT/B3LYP/6-311++G(d,p) method.
According to the literature [39], absorption band for pyridine compound was appeared at 210 nm with 3.0 eV. By the introduction of N in the ring the pyridine was formed to be pyrimidine and this form of molecule showed the E-band shift to the higher region. In addition to that, the substitution of electron with drawing group (amine) in the ring dislocated the UV-Visible band to the higher wavelength side at 255 nm minimum and 311 nm maximum. This form of bathochromic shift usually observed to indicate that, the compound possessed the rich biological property as well [40]. Here, from this appearance of peak, it was concluded that, the title molecule having enriched biological property.
Molecular electrostatic potential (MESP) map interpretation
The Electrostatic potential map is the charge gradient pictorial diagram for displaying spherically symmetric arrangements of electron clouds against the protonic content on their polar and non-polar species in the presence of electric field. The ability of the nuclei of the molecule to attract the electron clouds which are varied with respect to inter molecular effective dispersion forces existed among the atoms [41]. This type of forces induced the chemical potential called Molecular electrostatic potentials (MEPs) which generally made available the information regarding chemical reactivity or the biological activity of chemical compound.
The electrostatic potential map of the present molecule was depicted in between positive and negative potential contours in Figure 9. Here, the positive and negative potential gradient values were dispersed to be ± 8.117 e-2. From the figure, it was seen that, the positive attractive boundary called nucleophilic region appeared around H of amine group at top moiety of molecule. This view was showed that, due to the intake of electron cloud by the ring from amine group, the protonic content were incremented abnormally itself. So, the region appeared as blue which represent the nucleophilic region. The dislocated electron cloud was found to be accumulated over the imine groups in the ring which demonstrate the boundary of electrophilic region. The intermediate electrostatic potential was identified to be around methyl groups at the bottom moiety of the molecule. This area was observed to be green also called electron cloud migration region. The electropolarized isosurface was displayed in the Figure 9 and from the figure, it was found that, the field was dispersed over the compound at certain extent and this type of field illustrated electronic field scattering. This pyramidal type of charge separation showed the uniqueness of the biological activity.
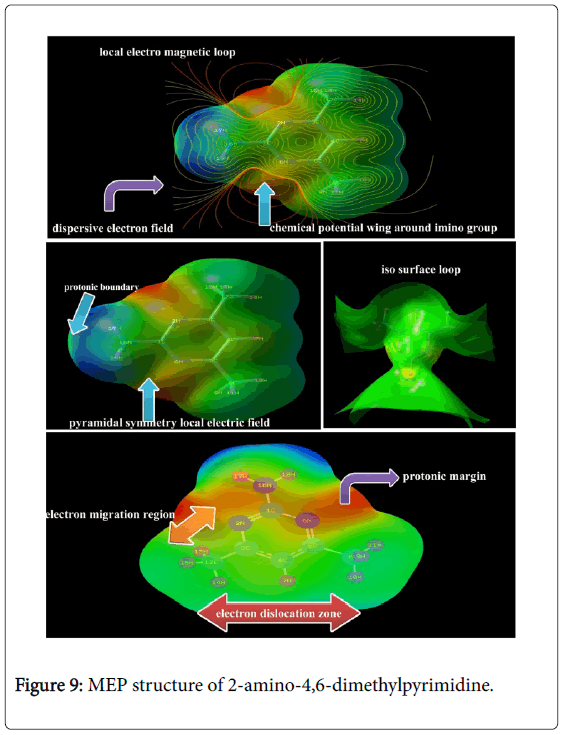
Figure 9: MEP structure of 2-amino-4,6-dimethylpyrimidine.
Physico-chemical properties
The intramolecular interaction profile is truly explicited the actual energy profile of the reacted compound. The excited energies among the atomic sites split the energy levels in order to accomplish equilibrium forces of attraction and repulsion. The energy separation at interior cascade orbitals showed the used chemical potential of compound to become drug. From that electronic energy of the orbitals, the physical and chemical parameters can be determined. The calculated parameters with specific measurement unit were depicted in the Table 7. These predicted chemical properties were extracted from parameters which are calculated from available bio-kinetic energies.
| Parameter | IR region | UV-Visible region | Electrophilicity charge transfer (ECT) (ΔNmax)A-(ΔNmax)B |
|---|---|---|---|
| Etotal (Hartree) | -398.46 | -398.28 | 3.01 |
| EHOMO (eV) | 6.344 | 6.558 | |
| ELUMO (eV) | 1.003 | 1.257 | |
| ΕΗ≅Μ≅−ΛΥΜ≅ γαπ (ες) | 5.341 | 5.301 | |
| EHOMO-1 (eV) | 6.83 | 6.763 | |
| ELUMO+1 (eV) | 0.277 | 0.517 | |
| ΕΗ≅Μ≅−1−ΛΥΜ≅+1 γαπ (ες) | 6.553 | 6.245 | |
| Chemical hardness (η) | 2.67 | 2.65 | |
| Electronegativity (χ) | -3.673 | -3.9 | |
| Chemical potential (μ) | -3.673 | -3.908 | |
| Chemical softness(S) | 0.187 | 0.188 | |
| Electrophilicity index(ω) | 2.526 | 2.881 | |
| Dipole moment | 1.096 | 2.294 |
Table 7: Physico-chemical parameters of 2-Amino-4, 6- Dimethylpyrimidine.
The molecular binding energy of -398.46 was utilized to fabricate the compound; 2-Amino-4, 6-Dimethylpyrimidine and it is very minimum by which three chemical entities were composed. The dipole moment is the measuring rate of charge dispersion in organic compound and if it is greater 1.0 dyne, it will be biologically very reactive. Accordingly, the measured dipole moment of the present case was 1.096 dyne in IR and 2.294 dyne in UV-Visible region. Hence, this compound was found to be much stable in electronic spectral region than vibrational region. This view showed the present compound is biologically reactive and the result was supported by the literature [42].
The coulomb reactive energy index of chemical bonds for executing intra molecular interactions is usually deliberated by the ionization potential [43]. The ionization potential was observed for this case 1.003 and 1.257 in IR and UV-Visible region respectively and it is greater than unity which was able to have moderate to make chemical stability reactive energy in molecular site.
The Electronegativity of aromatic compound is very significant factor for always used for measuring local electro-chemical reliability for the prediction of centre of magnetic polarity gradient of aromatic compound [44]. Here, it was measured to be 3.673 and 3.900 in IR and UV region respectively. The experiential value in both regions was extremely high and sufficient to connect with protein complex. It also established that, the spontaneous accretion of electron density to adopt very good drug action. This electro-magneto effect of molecular geometry was induced by π-bond (C=N) interacted orbitals of symmetrical imine groups of pyrimidine ring and was meaningfully ensured by MEP diagram.
The electrophilicity index is basically used to evaluate the rate of flow of potential energy through degenerate cascading molecular orbitals. In this case, the electrophilicity index in IR and UV-Visible region were 2.526 and 2.881 eV respectively. The present compound is the combination of pyrimidine and methyl coupled amino group and it was obviously high which demonstrated that, the enormous quantity of chemical energy was spontaneously generated in the ring with the help of amino group. This process was approved by observing the HOMO and LUMO electronic orbitals arrangement in Figure 7. The energy exchange was visibly evidenced from the ele
Conclusions
The fundamental physico-chemical properties related to the stable structure of present case; 2-amino-4,6-dimethylpyrimidine were interpreted. The active compositional parts for causing medical activity of compound were deeply analyzed and the property driving mechanism was determined. The biological and molecular properties were evaluated and intensively investigated towards drug property. Lipinski parameters were calculated and tested for the evaluation of antibiotic property. The reasoning of drug applications was determined from frontier molecular orbitals profile and energy transitions among electronic orbitals causing drug mechanism were identified. The chemical contamination test was carried out by VCD profile analysis and the substance under study was found to be having less toxicity effect.
References
- Brown DJ (1984) Eight-membered and larger Heterocyclic Rings and their Fused Derivatives, Other Seven-membered Rings. In: Katritzky AR, Rees CW (editors) Comprehensive Heterocyclic Chemistry (1stedn), Pergamon Press, Oxford, UK.
- Elderfield RC (1957) Heterocyclic Compounds. John Wiley & Sons, New York, USA.
- Ju Y, Varma RS (2006) Aqueous N-heterocyclization of primary amines and hydrazines with dihalides: microwave-assisted syntheses of N-azacycloalkanes, isoindole, pyrazole, pyrazolidine, and phthalazine derivatives. J Org Chem 71: 135-141.
- Ju Y, Kumar D, Varma RS (2006) Revisiting nucleophilic substitution reactions: microwave-assisted synthesis of azides, thiocyanates, and sulfones in an aqueous medium. J Org Chem 71: 6697-6700.
- Jeu L, Piacenti FJ, Lyakhovetskiy AG, Fung HB (2003) Voriconazole. Clin Therapeutics 25: 1321-1381.
- Poole K (2001) Overcoming antimicrobial resistance by targeting resistance mechanisms. J Pharm Pharmacol 53: 283-294.
- Ito S, Masuda K, Kusano S (1991) Pyrimidine derivative, process for preparing same and agricultural or horticultural fungicidal composition containing same. U.S. Patent, 4 (988), 704.
- Agarwal N, Raghuwanshi SK, Upadhyay DN, Shukla PK, Ram VJ (2000) Suitably functionalised pyrimidines as potential antimycotic agents. Bioorganic & Med Chem Lett 10: 703-706.
- Basavaraja HS, Sreenivasa GM, Jayachandran E (2005) Synthesis and biological activity of novel pyrimidino imidazolines. Indian J Heterocyclic Chem 15: 69.
- Coates AR, Hu Y (2007) Novel approaches to developing new antibiotics for bacterial infections. British J Pharmacol 152: 1147-1154.
- Moorthy N, Prabakar PJ, Ramalingam S, Govindarajan M, Gnanamuthu SJ, et al. (2016) Spectroscopic analysis, AIM, NLO and VCD investigations of acetaldehyde thiosemicarbazone using quantum mechanical simulations. J Physics Chem Solids 95: 74-88.
- Xavier S, Periandy S (2015) Spectroscopic (FT-IR, FT-Raman, UV and NMR) investigation on 1-phenyl-2-nitropropene by quantum computational calculations. Spectrochimi Acta A Mol Biomol Spectrosc 149: 216-230.
- Madanagopal A, Periandy S, Gayathri P, Ramalingam S, Xavier S (2017) Molecular structure activity on pharmaceutical applications of Phenacetin using spectroscopic investigation. J Mol Struct 1127: 611-625.
- Minkin VI, Glukhovtsev MN, Simkin BY (1994) Aromaticity and Antiaromaticity: Electronic and Structural Aspects (4th edn). Wiley, New York.
- Quesada A, Marchal A, Melguizo M, Low JN, Glidewell C (2004) Symmetrically 4, 6-disubstituted 2-aminopyrimidines and 2-amino-5-nitrosopyrimidines: interplay of molecular, molecular–electronic and supramolecular structures. Acta Crystallogr B Struct Sci 60: 76-89.
- Madanagopal A, Periandy S, Gayathri P, Ramalingam S, Xavier S (2017) Spectroscopical and Computational investigation on Pharmacological activity on the structure of 1-Benzylimidazole. J Taibah Univ Sci 11: 975-996.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews 23: 3-25.
- Lipinski CA (2004) Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today: Technologies 1: 337-341.
- Leo A, Hansch C, Elkins D (1971) Partition coefficients and their uses. Chem Rev 71: 525-616.
- Leeson PD, Springthorpe B (2007) The influence of drug-like concepts on decision-making in medicinal chemistry. Nat Rev Drug Discov 6: 881-890.
- Khoshneviszadeh M, Shahraki O, Khoshneviszadeh M, Foroumadi A, Firuzi O, et al. (2016) Structure-based design, synthesis, molecular docking study and biological evaluation of 1, 2, 4-triazine derivatives acting as COX/15-LOX inhibitors with anti-oxidant activities. J Enzyme Inhib Med Chem 31: 1602-1611.
- Prabhu T, Periandy S, Ramalingam S (2011) FT-IR and FT-Raman investigation, computed vibrational intensity analysis and computed vibrational frequency analysis on m-Xylol using ab-initio HF and DFT calculations. Spectrochimi Acta A Mol Biomol Spectrosc 79: 948-955.
- Varsányi G (1974) Assignments for vibrational spectra of seven hundred benzene derivatives. Halsted Press.
- Subramanian MK, Anbarasan PM, Manimegalai S (2009) DFT simulations and vibrational analysis of FT-IR and FT-Raman spectra of 2, 4-diamino-6-hydroxypyrimidine. Spectrochi Acta A Mol Biomol Spectrosc 73: 642-649.
- Peesole RL, Shield LD, McWillam IC (1976) Modern Methods of Chemical Analysis, Wiley, New York.
- Krishnakumar V, Balachandran V (2005) DFT studies, vibrational spectra and conformational stability of 4-hydroxy-3-methylacetophenone and 4-hydroxy-3-methoxyacetophenone. Spectrochimi Acta A Mol Biomo Spectrosc 61: 2510-2525.
- Xavier RJ, Balachandran V, Arivazhagan M, Ilango G (2010) Vibrational spectral analysis of 1-methoxynaphthalene. Indian J Pure Appl Physs 48: 245-250.
- Goel RK, Gupta SK, Agarwal ML, Sharma SN (1981) Infrared-Spectra of Some N-Heterocyclic Molecules of Biological Interest. Indian J Pure Appl Physs 19): 501-4.
- Susi H, Ard JS (1974) Planar valence force constants and assignments for pyrimidine derivatives. Spectrochimi Acta A Mol Spectrosc 30: 1843-1853.
- Ardyukova AF (1974) Atias of Spectra of Aromatic and Hetrocyclic Compounds, No.4, Infrared Spectra ofPyrimidine Series, Nauka Sib. Otd., Novosibirsk.
- Billes F, Endrédi H, Jalsovszky G (1999) Vibrational spectroscopy of diazoles. J Mol Struct: Theochem 465: 157-172.
- Krueger PJ, Thompson HW (1957) Vibrational Band Intensities in Substituted Anilines. In Proceedings of the Royal Society of London A: Mathematical, Physical and Engineering Sciences 243: 143-153.
- Krueger PJ (1962) Fundamental NH2 Stretching Frequencies in Amines and Amides. Nature 194: 1077-1078.
- Ohno K, Mandai Y, Matsuura H (1992) Vibrational spectra and molecular conformation of taurine and its related compounds. J Mol Struct 268: 41-50.
- Durig JR, Beshir WB, Godbey SE, Hizer TJ (1989) Raman and infrared spectra, conformational stability and Ab initio calculations for nâ€propylamine. J Raman Spectrosc 20: 311-333.
- Socrates G (2001) Infrared and Raman characteristic group frequencies: tables and charts. John Wiley & Sons.
- Armarego W, Katritzky AR, Ridgewell BJ (1964) The infra-red spectra of polycyclic heteroaromatic compounds—IV [1,2]: Monosubstituted quinazolines. Spectrochimi Acta 20: 593-596.
- Dhankar RP, Rahatgaonkar AM, Chorghade MS, Tiwari A (2012) Spectral and in vitro antimicrobial properties of 2-oxo-4-phenyl-6-styryl-1, 2, 3, 4-tetrahydro-pyrimidine-5-carboxylic acid transition metal complexes. Spectrochimi Acta A Mol Biomol Spectrosc 93: 348-353.
- Al-Hashimi NA, Hussein YH (2010) Ab initio study on the formation of triiodide CT complex from the reaction of iodine with 2, 3-diaminopyridine. Spectrochimi Acta A Mol Biomol Spectrosc 75: 198-202.
- Vektariene A, Vektaris G, Svoboda J (2009) A theoretical approach to the nucleophilic behavior of benzofused thieno [3, 2-b] furans using DFT and HF based reactivity descriptors. ARKIVOC: Online J Org Chem 2009: 311-329.
- Chang R (2005) Chapter 13: Physical chemistry for the biosciences. University Science Books.
- Cade PE, Bader RF, Henneker WH, Keaveny I (1969) Molecular Charge Distributions and Chemical Binding. IV. The Secondâ€Row Diatomic Hydrides AH. J Chem Phys 50: 5313-5333.
- Bader RF, Henneker WH, Cade PE (1967) Molecular charge distributions and chemical binding. J Chem Phy 46 3341-3363.
- Nivaldo TJ (2008) Chemistry: A Molecular Approach, 2nd Edn. New Jersey.
- Glendening ED, Reed AE, Carpenter JE, Weinhold F (1988) NBO, version 3.1; University of Wisconsin: Madison, WI.
- Hobza P, Havlas Z (2000) Blue-shifting hydrogen bonds. Chem Rev 100: 4253-4264.
- Paul BK, Mahanta S, Singh RB, Guchhait N (2010) A DFT-based theoretical study on the photophysics of 4-hydroxyacridine: single-water-mediated excited state proton transfer. The J Phys Chem A 114: 2618-2267.
Citation: Johnson P, George G, Ramalingam S, Periandy S (2018) Spectroscopic and QSAR analysis on Antibiotic drug; 2-amino-4,6- dimethylpyrimidine using Quantum Computational Tools. J Mol Pharm Org Process Res 6: 142. DOI: 10.4172/2329-9053.1000142
Copyright: © 2018 Johnson P, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 6311
- [From(publication date): 0-2018 - Dec 08, 2025]
- Breakdown by view type
- HTML page views: 5253
- PDF downloads: 1058