Research Article Open Access
UV-Visible Spectrophotometric Method Development and Validation of Assay of Paracetamol Tablet Formulation
Siladitya Behera*, Subhajit Ghanty, Fahad Ahmad, Saayak Santra, and Sritoma Banerjee
Department of Quality Assurance and Pharma Regulatory Affairs, Gupta College of Technological Sciences, West Bengal, India
- *Corresponding Author:
- Siladitya Behera
Department of Quality Assurance and Pharma Regulatory Affairs
Gupta College Of Technological Sciences
Ashram More, G.T.Road
Asanol-713301, Burdwan District
West Bengal, India
E-mail: Siladitya.behera@gmail.com
Received date: October 01, 2012; Accepted date: October 27, 2012; Published date: October 31, 2012
Citation: Behera S, Ghanty S, Ahmad F, Santra S, Banerjee S (2012) UV-Visible Spectrophotometric Method Development and Validation of Assay of Paracetamol Tablet Formulation. J Anal Bioanal Tech 3:151. doi: 10.4172/2155-9872.1000151
Copyright: © 2012 Behera S, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Analytical & Bioanalytical Techniques
Abstract
A novel,safe and sensitive method of spectrophotometric estimation in UV-region has been developed for the assay of Paracetamol in its tablet formulation. The method have been developed and validated for the assay of Paracetamol using Methanol and water as diluents. Which does not shows any interference in spectrophotometric estimations. All the parameters of the analysis were chosen according to ICH [Q2(R1)] guideline and validated statistically using RSD and %RSD along with neat chromate grams.
Keywords
Spectrophotometric; Developed; Validated; Parameters; ICH[Q2(R1)]
Introduction
It is the branch of science dealing with the study of interaction between Electromagnetic radiation and matter. It is a most powerful tool available for the study of atomic and molecular structure/s and is used in the analysis of wide range of samples. Optical spectroscopy includes the region on electromagnetic spectrum between 100 Å and 400 μm. The regions of electromagnetic spectrum are shown in table 1.
| Region | Wavelength |
|---|---|
| Far (or vacuum)ultraviolet | 10-200 nm |
| Near ultraviolet | 200-400 nm |
| Visible | 400-750 nm |
| Near infrared | 0.75- 2.2 μm |
| Mid infrared | 2.5-50 μm |
| Far infrared | 50-1000 μm |
Table 1: Regions of electromagnetic spectrum.
Ultraviolet-Visible spectrophotometry [3]
UV-Visible spectrophotometry is one of the most frequently employed technique in pharmaceutical analysis. It involves measuring the amount of ultraviolet or visible radiation absorbed by a substance in solution. Instrument which measure the ratio, or function of ratio, of the intensity of two beams of light in the U.V-Visible region are called Ultraviolet-Visible spectrophotometers.
In qualitative analysis, organic compounds can be identified by use of spectrophotometer, if any recorded data is available, and quantitative spectrophotometric analysis is used to ascertain the quantity of molecular species absorbing the radiation. Spectrophotometric technique is simple, rapid, moderately specific and applicable to small quantities of compounds. The fundamental law that governs the quantitative spectrophotometric analysis is the Beer -Lambert law.
Beer’s law: It states that the intensity of a beam of parallel monochromatic radiation decreases exponentially with the number of absorbing molecules. In other words, absorbance is proportional to the concentration.
Lambert’s law: It states that the intensity of a beam of parallel monochromatic radiation decreases exponentially as it passes through a medium of homogeneous thickness. A combination of these two laws yields the Beer-Lambert law.
Beer-Lambert law: When beam of light is passed through a transparent cell containing a solution of anabsorbing substance, reduction of the intensity of light may occur. Mathematically, Beer- Lambert law is expressed as
A=a b c
Where, A=absorbance or optical density
a=absorptivity or extinction coefficient
b=path length of radiation through sample (cm)
c=concentration of solute in solution.
Both b and a are constant so a is directly proportional to the concentration c
When c is in gm/100 ml, then the constant is called A (1%, 1 cm)
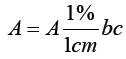
Quantification of medicinal substance using spectrophotometer may carried out by preparing solution in transparent solvent and measuring it’s absorbance at suitable wavelength. The wavelength normally selected is wavelength of maximum absorption (λmax), where small error in setting the wavelength scale has little effect on measured absorbance. Ideally, concentration should be adjusted to give an absorbance of approximately 0.9, around which the accuracy and precision of the measurements are optimal.
The assay of single component sample, which contains other absorbing substances, is then calculated from the measured absorbance by using one of three principal procedures. They are, use of standard absorptivity value, calibration graph and single or double point standardization. In standard absorptive value method, the use of standard A (1%, 1 cm) or E values are used in order to determine its absorptivity. It is advantageous in situations where it is difficult or expensive to obtain a sample of the reference substance. In calibration graph method, the absorbances of a number of standard solutions of the reference substance at concentrations encompassing the sample concentrations are measured and a calibration graph is constructed. The concentration of the analyte in the sample solution is read from the graph as the concentration corresponding to the absorbance of the solution. The single point standardization procedure involves the measurement of the absorbance of a sample solution and of a standard solution of the reference substance. The concentration of the substances in the sample is calculated from the proportional relationship that exists between absorbance and concentration.
Ctest= (Atest×Cstd)/Astd
Where Ctest and Cstd are the concentrations in the sample and standard solutions respectively and Atest and Astd are the absorbances of the sample and standard solutions respectively. For assay of substance/s in multi component samples by spectrophotometer; the following methods are being used routinely, which includes [4],
• Simultaneous equation method
• Derivative spectrophotometric method
• Absorbance ratio method (Q-Absorbance method)
• Difference spectrophotometry
• Solvent extraction method
Introduction to Paracetamol
Paracetamol or acetaminophen is a widely used over-the-counter analgesic (pain reliever) and antipyretic (fever reducer). It is commonly used for the relief of headaches and other minor aches and pains and is a major ingredient in numerous cold and flu remedies. In combination with opioid analgesics, Paracetamol can also be used in the management of more severe pain such as post-surgical pain and providing palliative care in advanced cancer patients [5]. The onset of analgesia is approximately 11 minutes after oral administration of Paracetamol [6], and its half-life is 1-4 hours. Though acetaminophen is used to treat inflammatory pain, it is not generally classified as an NSAID because it exhibits only weak anti-inflammatory activity.
While generally safe for use at recommended doses (1,000 mg per single dose and up to 4,000 mg per day for adults), acute overdoses of Paracetamol can cause potentially fatal liver damage and, in rare individuals, a normal dose can do the same; the risk is heightened by alcohol consumption. Paracetamol toxicity is the foremost cause of acute liver failure in the Western world, and accounts for most drug overdoses in the United States, the United Kingdom, Australia and New Zealand [7-10].
It is the active metabolite of Phenacetin, once popular as an analgesic and antipyretic in its own right, but unlike Phenacetin and its combinations, Paracetamol is not considered carcinogenic at therapeutic doses [11]. The words acetaminophen (used in the United States, Canada, Japan, South Korea, Hong Kong, and Iran [12] and Paracetamol (used elsewhere) both come from a chemical name for the compound: para-acetylaminophenol and para-acetylaminophenol. In some contexts, it is simply abbreviated as APAP, for acetyl-paraaminophenol.
Description
Systematic (IUPAC) name:N-(4-hydroxyphenyl)acetamide
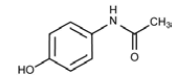
Paracetamol is part of the class of drugs known as “aniline analgesics”; it is the only such drug still in use today [13]. It is not considered an NSAID because it does not exhibit significant antiinflammatory activity (it is a weak COX inhibitor) [14,15]. This is despite the evidence that Paracetamol and NSAIDs have some similar pharmacological activity [16].
Mechanism of action
To date, the mechanism of action of Paracetamol is not completely understood. The main mechanism proposed is the inhibition of cyclooxygenase (COX), and recent findings suggest that it is highly selective for COX-2 [17]. While it has analgesic and antipyretic properties comparable to those of aspirin or other NSAIDs, its peripheral anti-inflammatory activity is usually limited by several factors, one of which is the high level of peroxides present in inflammatory lesions. However, in some circumstances, even peripheral anti-inflammatory activity comparable to other NSAIDs can be observed. An article [18] in Nature Communications from researchers in London, UK and Lund, Sweden in November 2011 has found a hint to the analgesic mechanism of acetaminophen (Paracetamol), being that the metabolites of acetaminophen e.g. NAPQI, act on TRPA1-receptors in the spinal cord to suppress the signal transduction from the superficial layers of the dorsal horn, to alleviate pain.
Metabolism
Paracetamol is metabolised primarily in the liver, into non-toxic products. Three metabolic pathways are no table:
• Glucuronidation is believed to account for 40% to two-thirds of the metabolism of paracetamol.
• Sulfation (sulfate conjugation) may account for 20-40% [19].
• N-hydroxylation and rearrangement, then GSH conjugation, accounts for less than 15%. The hepatic cytochrome P450 enzyme system metabolizes paracetamol, forming a minor yet significant alkylating metabolite known as NAPQI (N-acetyl-p-benzo-quinone imine). NAPQI is then irreversibly conjugated with the sulfhydryl groups of glutathione [20].
All three pathways yield final products that are inactive, nontoxic, and eventually excreted by the kidneys. In the third pathway, however, the intermediate product NAPQI is toxic. NAPQI is primarily responsible for the toxic effects of Paracetamol; this constitutes an example of toxication.
Method validation
• Validation is concerned with assuring that a measurement process produces valid measurements.
• Results from method validation can be used to judge the quality, reliability and consistency of analytical results. It is an integral part of any good analytical practice.
• A measurement process producing valid measurements for an intended application is fit for purpose.
Method validation is the process used to confirm that the analytical procedure employed for a specific test is suitable for its intended use. Results from method validation can be used to judge the quality, reliability and consistency of analytical results; it is an integral part of any good analytical practice.
Analytical methods need to be validated or revalidated,
• Before their introduction into routine use;
• Whenever the conditions change for which the method has been validated (e.g., an instrument with different characteristics or samples with a different matrix); and
• Whenever the method is changed and the change is outside the original scope of the method.
Nowadays, there are several international renowned organisations offering guidelines on method validation and related topics.
• American Society for Testing and Material (ASTM)
• Codex Committee on Methods of Analysis and Sampling (CCMAS)
• European Committee for Normalization (CEN)
• Cooperation on International Traceability in Analytical Chemistry (CITAC)
• European Cooperation for Accreditation (EA)
• Food and Agricultural Organization (FAO)
• United States Food and Drug Administration (FDA)
• International Conference on Harmonization (ICH).
ICH Guidelines (ICH Q2R1) for Analytical Procedure and Validation [21]
The analytical procedure refers to the way of performing the analysis. It should describe in detail the steps necessary to perform each analytical test. This may include but is not limited to: the sample, the reference standard and the reagents preparations, use of the apparatus, generation of the calibration curve, use of the formula for the calculation, etc.
Types of Analytical Procedures to be validated
The discussion of the validation of analytical procedures is directed to the four most common types of analytical procedures:
- Identification tests;
- Quantitative tests for impurities’ content;
- Limit tests for the control of impurities;
- Quantitative tests of the active moiety in samples of drug substance or drug product or other selected component(s) in the drug product.
The objective of the analytical procedure should be clearly understood since this will govern the validation characteristics which need to be evaluated. Typical validation characteristics which should be considered are listed below:
• Accuracy
• Precision
• Repeatability
• Intermediate Precision
• Specificity
• Detection Limit
• Quantitation Limit
• Linearity
• Range
Furthermore revalidation may be necessary in the following circumstances:
-Changes in the synthesis of the drug substance;
- Changes in the composition of the finished product;
- Changes in the analytical procedure.
Aim of Present Work
This work deals with the validation of the developed method for the assay of Paracetamol from its dosage form (tablets). Hence, the method can be used for routine quality control analysis and also stability.
The aim and scope of the proposed work are as under:
• To develop suitable spectrophotometric method for assay of Paracetamol tablet.
• Perform the validation for the method.
Experimental
Materials
Paracetamol standard of was provided by Torque Pharmaceuticals (P) Ltd. (India). Paracetamol tablets containing 500 mg Paracetamol and the inactive ingredient used in drug matrix were obtained from market. Analytical grade methanol and water were obtained from Spectrochem Pvt. Ltd., Mumbai (India).
Diluent preparation
Methanol and water (15:85, v/v) used as a diluent.
Standard preparation
10 mg drug was dissolved in 15 ml methanol and was shaken well. Then 85 ml water was added to it to adjust the volume up to 100 ml (100 ppm). From that 5 ml was taken and volume was adjusted up to 50 ml with diluents.
Test preparation
20 tablets were weighed and powdered. Powdered tablet equivalent to 100 mg of paracetamol was weighed and taken into 100 ml volumetric flask then 15 ml of methanol was added and shaken well to dissolve it after that 85 ml of water was added to adjust the volume up to 100 ml. From that 1 ml of solution was withdrawn and taken in 100 ml volumetric flask. The volume was adjusted with diluent up to 100 ml.
Instrumentation
UV-Visible double beam spectrophotometer with matched quartz cells (1 cm)
Model: Evolution 201
Make: Thermo Scientific, 81 Wyman Street Waltham, Massachusetts, US.
Results and Discussion
Development and optimization of the spectrophotometric method
Proper wave length selection of the methods depends upon the nature of the sample and its solubility. To develop a rugged and suitable spectrophotometric method for the quantitative determination of paracetamol, the analytical condition were selected after testing the different parameters such as diluents, buffer, buffer concentration, and other chromatographic conditions.
Our preliminary trials were by using different compositions of diluents consisting of water with buffer and methanol. By using diluent consisted of methanol - water (50:50, v/v) best result was obtained and degassed in an ultrasonic bath (Enertech Electronics Private Limited). Below figures represent the spectrums of blank, standard and test preparation respectively.
Selection of wavelength
Scan standard solution in UV spectrophotometer between 200 nm to 400 nm on spectrum mode, using diluents as a blank (Figure 1).

Figure 1: UV spectrum of Paracetamol in Standard solution.
Paracetamol shows λmax at 243. The proposed analytical method is simple, accurate and reproducible (Figure 2).

Figure 2: UV spectrum of Paracetamol in Sample solution.
Method validation
Specificity: Resolution of the analyte peak from the nearest peak: Solution of each of the analyte was injected separately and their retention time is noted. The standard working solution containing a mixture of the component being analyze is also injected and each of analyte peaks is check for its resolution from the nearest (Figures 2,3,4,5).

Figure 3: UV Spectrum of Placebo.

Figure 4: UV Spectrum of Placebo+analyte.

Figure 5: UV Spectrum of Blank.

Figure 6: Linearity curve for Paracetamol.
Linearity: Six points calibration curve were obtained in a concentration range from 0-150 ppm for Paracetamol. The response of the drug was found to be linear in the investigation concentration range and the linear regression equation was y = 0.004x+0.007 with correlation coefficient 0.998 (Table 2, Figure 6).
| Conc(ppm) | Absorbance |
|---|---|
| 0 | 0 |
| 50 | 0.246 |
| 75 | 0.338 |
| 100 | 0.456 |
| 125 | 0.582 |
| 150 | 0.672 |
Table 2: Conc. Vs Abs. table for Linearity Study.
Precision: Precision of the analytical method is ascertained by carrying out the analysis as per the procedure and as per normal weight taken for analysis. Repeat the analysis six times. Calculate the % assay, mean assay, % Deviation and % relative standard deviation and %RSD.
The developed method was found to be precise as the %RSD values for the repeatability and intermediate precision studies were <0.98% and <0.79%, respectively (Table 3).
| Sample No. | % Assay | |
|---|---|---|
| SET | Intraday | Interday |
| 1 | 101.8 | 99.3 |
| 2 | 101.2 | 98.2 |
| 3 | 101.3 | 99.0 |
| 4 | 101.5 | 99.0 |
| 5 | 100.6 | 98.6 |
| 6 | 99.1 | 100.5 |
| Mean | 101.1 | 99.1 |
| SD | 0.99 | 0.78 |
| %RSD | 0.98 | 0.79 |
Table 3: Evaluation data of precision study.
Accuracy: Accuracy of the method is ascertained by standard addition method at 3 levels. Standard quantity equivalent to 50%, 100% and 125% is to be added in sample. The result shown that best recoveries (98.54-99.13%) of the spiked drug were obtained at each added concentration, indicating that the method was accurate (Table 4). Composition of Placebo Added for Accuracy Study is shown in Table 5.
| % Recovery Level | % Recovery | Mean %Recovery | SD | %RSD |
|---|---|---|---|---|
| 50% | 98.62 | 98.62 | 0 | 0 |
| 98.63 | 0.0057735 | 0.005854 | ||
| 98.61 | 0.00816497 | 0.008279 | ||
| 100% | 98.56 | 98.55 | 0.00693889 | 0.007041 |
| 98.57 | 0.00942809 | 0.009566 | ||
| 98.54 | 0.1503083 | 0.015251 | ||
| 125% | 99.13 | 99.12 | 0.00707107 | 0.007134 |
| 99.11 | 0.00707107 | 0.007134 | ||
| 99.12 | 0 | 0 |
Table 4: Evaluation data of accuracy study.
| Time (Hrs.) | Standard | Sample |
|---|---|---|
| 0 | 0.723 | 0.723 |
| 2 | 0.72 | 0.721 |
| 4 | 0.717 | 0.717 |
| 6 | 0.716 | 0.717 |
| 8 | 0.716 | 0.716 |
| Limit at (2 hr) | 0.414 | 0.276 |
| Limit at (4 hr) | 0.829 | 0.829 |
| Limit at (6 hr) | 0.968 | 0.829 |
| Limit at (8 hr) | 0.968 | 0.968 |
Table 5: Evaluation data of solution stability study.
Solution stability study: Table 6 shows the results obtain in the solution stability study at different time intervals for test preparation. It was concluded that the test preparation solution was found stable up to 8 hr at room temperature, as during this time the result was not decrease below the minimum percentage.
| Sl no. | 242 nm | 243 nm | 244 nm |
|---|---|---|---|
| 1 | 0.65 | 0.653 | 0.657 |
| 2 | 0.65 | 0.653 | 0.656 |
| 3 | 0.651 | 0.653 | 0.656 |
| 4 | 0.651 | 0.653 | 0.657 |
| 5 | 0.651 | 0.654 | 0.657 |
| 6 | 0.652 | 0.654 | 0.657 |
| Mean | 0.650833 | 0.6533333 | 0.6566667 |
| SD | 0.000753 | 0.0005164 | 0.0005164 |
| %RSD | 0.115663 | 0.0790405 | 0.0786393 |
Table 6: Evaluation data of robustness study.
Robustness: The evaluation of robustness should be considered during the development phase and depends on the type of procedure under study. It should show the reliability of an analysis with respect to deliberate variations in method parameters.
If measurements are susceptible to variation in analytical conditions, the analytical condition should be suitably controlled or a precautionary statement should be included in the procedure.
The result of robustness study of the developed assay method was established in Table 7. The result shown that during all variance conditions, assay value of the test preparation solution was not affected and it was in accordance with that of actual. System suitability parameters were also found satisfactory; hence the analytical method would be concluded as robust.
| Sample No. | Absorbance |
|---|---|
| 1 | 0.66 |
| 2 | 0.661 |
| 3 | 0.66 |
| 4 | 0.66 |
| 5 | 0.66 |
| 6 | 0.66 |
| Average | 0.66 |
| SD | 0.000408 |
| %RSD | 0.061856 |
Table 7: Evaluation data of System Suitability Study.
System suitability: A system suitability test of the spectrophotometric system was performed before each validation run. Six replicate reading of standard preparation were taken and %RSD of standard reading were taken for same. Acceptance criteria for system suitability, %RSD of standard reading not more than 2.0%, were full fill during all validation parameter (Table 7).
Conclusion
The present analytical method was validated as per ICH Q2(R1) guideline and it meets to specific acceptance criteria. It is concluded that the analytical method was specific, precise, linear, accurate, robust and having stability indicating characteristics. The present analytical method can be used for its intended purpose.
Calculation formula used
1. Calculation formula for % assay of Paracetamol
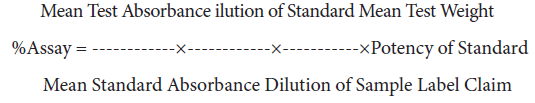

References
- Willard-Hobart H, Merritt Jr Lynne L, Dean John A (1974) Instrumental Methods of Analysis. (5thedn), Von Nostrand, University of Michigan.
- ChatwalGR, Anand S (2002) Instrumental Methods of Chemical Analysis. (5thedn), Himalaya Publishing House, New Delhi.
- Davidson AG (2002) Ultraviolet-visible absorption spectrophotometry. In Beckett AH, Stenlake JB, (4thedn), Practical Pharmaceutical chemistry. CBS Publishers and distributors, New Delhi, 275-278.
- Patil KM, Bodhankar SL (2005) High-performance thin-layer chromatographic determination of lamotrigine in serum. J Chromatogr B Analyt Technol Biomed Life Sci 823: 152-157.
- Scottish Intercollegiate Guidelines Network (SIGN) (2008) Control of pain in adults with cancer-A national clinical guideline. Scotland: National Health Service (NHS).
- Moller P, Sindet-Pedersen S, Petersen CT, Juhl GI, Dillenschneider A, et al. (2005) Onset of acetaminophen analgesia: comparison of oral and intravenous routes after third molar surgery. Br J Anaesth 94: 642-648.
- Daly FF, Fountain JS, Murray L, Graudins A, Buckley NA (2008) Guidelines for the management of paracetamol poisoning in Australia and New Zealand-explanation and elaboration. A consensus statement from clinical toxicologists consulting to the Australasian poisons information centres. Med J Aust 188: 296-301.
- Khashab M, Tector AJ, Kwo PY (2007) Epidemiology of acute liver failure. Curr Gastroenterol Rep 9: 66-73.
- Hawkins LC, Edwards JN, Dargan PI (2007) Impact of restricting paracetamol pack sizes on paracetamol poisoning in the United Kingdom: a review of the literature. Drug Saf 30: 465-479.
- Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, et al. (2005) Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 42: 1364-1372.
- Bergman K, Müller L, Teigen SW (1996) Series: current issues in mutagenesis and carcinogenesis, No. 65. The genotoxicity and carcinogenicity of paracetamol: a regulatory (re)view. Mutat Res 349: 263-288.
- Bradley N (1996) Naming of drugs. BMJ should use "paracetamol" instead of "acetaminophen" in its index. BMJ 313: 689.
- Bertolini A, Ferrari A, Ottani A, Guerzoni S, Tacchi R, et al. (2006) Paracetamol: new vistas of an old drug. CNS Drug Reviews 12: 250-275.
- Viswanathan AN, Feskanich D, Schernhammer ES, Hankinson SE (2008) Aspirin, NSAID, and Acetaminophen Use and the Risk of Endometrial Cancer. Cancer Res 68: 2507-2513.
- Altinoz MA, Korkmaz R (2004) NF-kappaB Macrophage migration inhibitory factor and cyclooxygenase-inhibitions as likely mechanisms behind the acetaminophen- and NSAID-prevention of the ovarian cancer. Neoplasma 51: 239-247.
- Byrant B, Knights K, Salerno E (2006) Pharmacology for health professionals. Mosby Elsevier.
- Hinz B, Cheremina O, Brune K (2008) Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. FASEB J22: 383-390.
- Andersson DA, Gentry C, Alenmyr L, Killander D, Lewis SE, et al. (2011) TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ(9)-tetrahydrocannabiorcol. Nat Commun 2: 551.
- Hendrickson RG, Kenneth EB (2006) Acetaminophen. In Flomenbaum NE, Goldfrank LR, Hoffman RS, Howland MA, Lewin NA, et al. Goldfrank's Toxicologic Emergencies 525.
- Borne Ronald F (1995) Nonsteroidal Anti-inflammatory Drugs. In Principles of Medicinal Chemistry, (4thedn), William OFoye, Thomas L Lemke, David A Williams, Williams & Wilkins, 544-545.
- ICH (1996) Q2B Validation of Analytical Procedures-Methodology. Consensus Guidelines, ICH Harmonized Tripartite Guidelines.
Relevant Topics
Recommended Journals
Article Tools
Article Usage
- Total views: 188449
- [From(publication date):
November-2012 - Dec 06, 2025] - Breakdown by view type
- HTML page views : 170231
- PDF downloads : 18218