 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Our Group organises 3000+ Global Conferenceseries Events every year across USA, Europe & Asia with support from 1000 more scientific Societies and Publishes 700+ Open Access Journals which contains over 50000 eminent personalities, reputed scientists as editorial board members.
Open Access Journals gaining more Readers and Citations
700 Journals and 15,000,000 Readers Each Journal is getting 25,000+ Readers
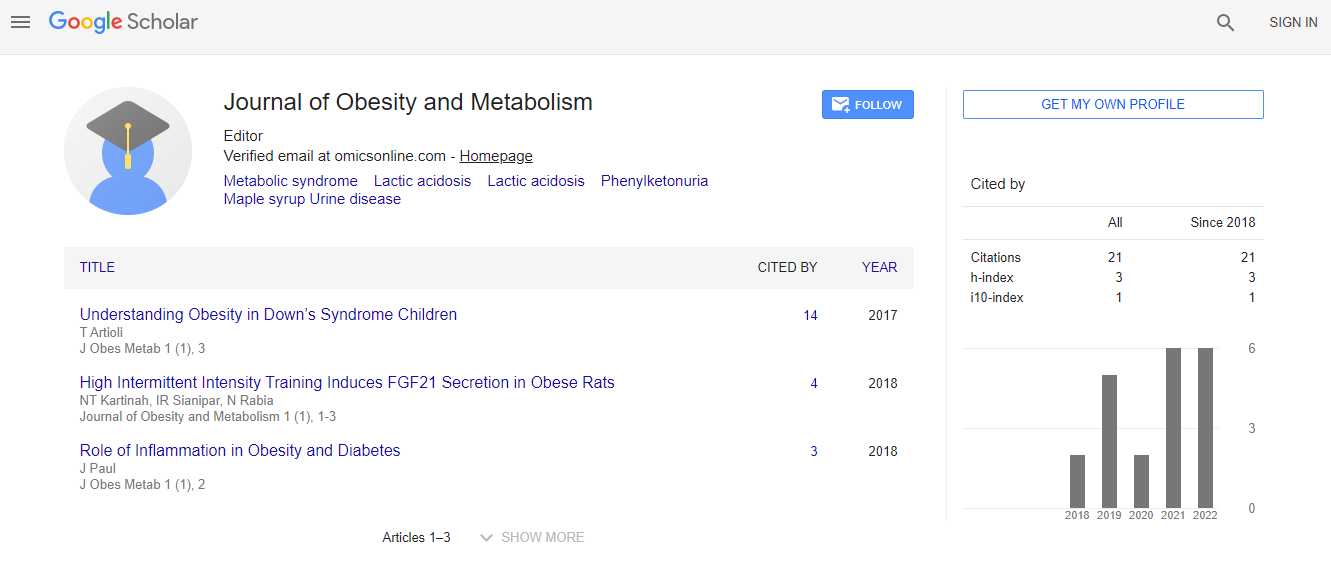
Google Scholar citation report
Citations : 24
Journal of Obesity and Metabolism received 24 citations as per Google Scholar report
Indexed In
- Google Scholar
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- Publons
- Euro Pub
- ICMJE
Useful Links
Share This Page

Editorial Board

Sylvie A. Akohoue, PhD, CNS
Adjunct Assistant Professor
Department of Medicine
Division of Gastroenterology
Meharry Medical College
Nashville, USA

Dr. Mahir Khalil Ibrahim Jallo
Clinical Professor of Medicine & Senior Consultant
Internal Medicine - Diabetes & Endocrinology
Gulf Medical University
United Arab Emirates

Jianfei Yu
Maple Respiratory Group
Canada

Honorary Professor of International Studies
School of Health Administration
Texas State University
USA
Submit Manuscript
Journal Impact Factor 0.37*
Submit manuscript at or send as an e-mail attachment to the Editorial Office at obesity@scienceresearchpub.org
If you are interested in publishing with us or have any questions, please feel free to contact us directly on WhatsApp .
Table of Contents
About the Journal
The Journal of Obesity and Metabolism is an interdisciplinary journal that aims at publishing any high quality, original research content that is related to obesity and also metabolism. The major focus of the journal is to bring awareness in the scientific community about all the recent developments and technological advancements in the diagnosis, management and even prevention of obesity which is a metabolic anomaly that is characterized by fat content accumulation in the body as a result of poor metabolism.
Obesity results in poor quality of life which is mainly because of the lack of ease in the movement due to the individual being overweight. It can be life threatening at times as obesity brings about various other metabolic conditions. It may also be a result of over consumption of food but may also be due to the genes from parents having these obesity genes. It is an interesting field of study as there is no proper basis for the statement that obesity is a resultant of poor metabolism, hence a lot of research is being conducted on finding the same.
The journal prioritises the publication of research that throws light on the new and existing therapies, including dietary-excerscise-lifestyle based interventions in the effective management and obesity as well as its related conditions. The journal follows a double blind peer review process which takes place rapidly and is finished in 21 days after which the articles are up for publishing. All the manuscripts based on obesity and metabolism along with any other related topics are welcome in the form of any type of articles which include but not limited to Short reviews, Commentaries, Research articles, Review articles, Short communications along with Letter to Editor etc.
Metabolic Acidosis
When too much acid is produced in the body of an individual, it results in metabolic acidosis. This can be the resultant of the kidneys not being able to function well to remove the acids from the system. Metabolic acidosis can be of many types namely: Diabetic ketoacidosis, hyperchloremic acidosis, distal renal tubular acidosis, proximal renal tubular acidosis, aspirin/methanol/ethylene glycol poisoning.
Metabolic syndrome
It is a set of conditions, mainly increased blood sugar, elevated blood pressure, excessive body fat specially around waist area, abnormal cholesterol levels, increased heart disease risk, stroke and diabetes.
Lactic acidosis
The majority of the lactic acid is produced in the muscle cells and red blood cells of an individual. This forms when the body breaks down carbohydrates to use for energy when oxygen levels are low. Lactic acidosis is a huge build-up of lactic acid in the body. This may be due to many conditions of which the main ones are Cancer, high alcohol consumption, vigorous exercise for long time, hypoglycaemia, liver failure, salicylates based medication, prolonged lack of oxygen, seizures, sepsis.
Hurler Syndrome
It is a type of disorder that ends up in a huge build-up of glycosaminoglycans due to the deficiency of alpha-L iduronidase, an enzyme that is responsible for degradation of mucoplysaccharides in the lysosomes. Symptoms may be visible early in the childhood and death may occur as a result of organ damage. This disease is autosomal recessive in nature.
Hunter syndrome
Hunter syndrome or mucopolysaccharidosis II can be termed as a lysosomal storage disease that is characterized by the deficiency of the enzyme iduronate-2-sulfatase. Heparan sulphate and dermatan sulphate are the accumulated substrates in Hunter syndrome. This condition has an X-linked recessive inheritance.
Heart disease and stroke
Excessive weight makes an individual more prone to a higher blood pressure as well as high levels of cholesterols. Both of these two make heart diseases or strokes inevitable.
Galactosemia
It is a rare genetic metabolic disorder that effects the ability of an individual to metabolise the sugar – galactose efficiently. This condition follows an autosomal recessive type of inheritance from the parents to the offspring that confers a deficiency in the enzyme that breaks down galactose. This condition is more common in the Irish Traveller population.
Porphyria
It is a type of hereditary disease that shows an abnormal metabolism of the blood pigment haemoglobin. Porphyrins are excreted via urine which turns darker , while other symptoms may also include metal disturbances and extreme sensitivity of the skin towards light.
Phenylketonuria
It is an inborn error of metabolism that ends up in reduced metabolism of phenylalanine- an amino acid. This condition can lead to intellectual disabilities, behavioural changes, seizures, mental disorders etc. It can also cause a musty smell and a lighter looking skin. Offspring born to poorly treated PKU inheritant mothers develop heart problems, smaller heads, and a very low birth weight.
Maple syrup Urine disease
It is a metabolic disease that is mainly characterized by poor processing of certain amino acids. The condition gets its name from the characteristic sweet odour of the affected infants’ urine. These children may also show symptoms like lethargy, delayed development, vomiting, poor feeding habits etc. This may also lead to coma, seizures or even death if left untreated.
Gaucher Disease
It is a condition in which an individual is not capable of producing the enzyme glucocerebrosidase that breaks down a fatty chemical in the system named glucocerebroside. Cells with excessive glucocerebroside accumulate in the liver/spleen of an individual which leads to the enlargement in the organs and is often painful. It also may interfere with completely having a meal. There is also reduced blood count, bleeding problems along with bone problems.
Journal Highlights
Fast Editorial Execution and Review Process (FEE-Review Process):
Journal of Obesity and Metabolism is participating in the Fast Editorial Execution and Review Process (FEE-Review Process) with an additional prepayment of $99 apart from the regular article processing fee. Fast Editorial Execution and Review Process is a special service for the article that enables it to get a faster response in the pre-review stage from the handling editor as well as a review from the reviewer. An author can get a faster response of pre-review maximum in 3 days since submission, and a review process by the reviewer maximum in 5 days, followed by revision/publication in 2 days. If the article gets notified for revision by the handling editor, then it will take another 5 days for external review by the previous reviewer or alternative reviewer.Acceptance of manuscripts is driven entirely by handling editorial team considerations and independent peer-review, ensuring the highest standards are maintained no matter the route to regular peer-reviewed publication or a fast editorial review process. The handling editor and the article contributor are responsible for adhering to scientific standards. The article FEE-Review process of $99 will not be refunded even if the article is rejected or withdrawn for publication.
The corresponding author or institution/organization is responsible for making the manuscript FEE-Review Process payment. The additional FEE-Review Process payment covers the fast review processing and quick editorial decisions, and regular article publication covers the preparation in various formats for online publication, securing full-text inclusion in a number of permanent archives like HTML, XML, and PDF, and feeding to different indexing agencies.
h-index
Articles published in Journal of Obesity and Metabolism have been cited by esteemed scholars and scientists all around the world. Journal of Obesity and Metabolism has got h-index 3, which means every article in Journal of Obesity and Metabolism has got 3 average citations.
Recently Published Articles
-
Self-efficacy Effects on Health effect and Body Weight
-
Self-Regulation and Management of Child Obesity
-
Weight Gain on Psychotropic Drugs: Has the Obesity Community been Paying Attention
-
Prevalence and Complications of Obesity in Surgical Patients: A Study in Benin, a Sub-Saharan African Country
-
Is The ?Health at Every Size? Approach Useful for Addressing Obesity