Disrupted Blood-CSF Barrier to Urea and Creatinine in Mild Cognitive Impairment and Alzheimer's Disease
Received: 20-Mar-2018 / Accepted Date: 30-Mar-2018 / Published Date: 07-Apr-2018 DOI: 10.4172/2161-0460.1000435
Abstract
Objective: In this pilot study hypothesizing that blood-CSF barrier (BCSFB) function is altered in mild cognitive impairment (MCI), we evaluated small-sized biomarker distribution between serum (SER) and cerebrospinal fluid (CSF). For both MCI and Alzheimer (AD) patients we quantified CSF neurochemistry; and compared CSF/SER ratios for urea and creatinine, as well as albumin, to those of healthy controls.
Methods: A compromised BCSFB in neurodegenerative states alters CSF-to-serum (CSF/SER) concentrations. We analyzed urea, creatinine and albumin, for transbarrier (across choroid plexus) distribution between CSF and serum, from patients with MCI (n=8) or AD (n=13). Lumbar CSF and arterial blood were frozen/analyzed by multiplex technology.
Results: In healthy controls, the CSF creatinine was significantly concentrated ~50% above the serum level. In both MCI and AD, the CSF creatinine concentration decreased while the urea level increased; CSF albumin was also elevated in AD. CSF/SER ratios for controls, MCI and AD were: urea 0.80, 0.98, 0.86; creatinine 1.52, 1.13, 1.14; and albumin 0.0045, 0.0051, 0.0065. Thus, CSF/SER ratios for creatinine and urea in MCI were similar to those in AD patients.
Conclusion: Blood-CSF barrier compromise in MCI resembled that in AD. In cognitively-impaired patients, the dissipating ratios toward equilibrium suggest disease-altered BCSFB permeability (urea and albumin) and transporter activity (creatinine/creatine). We propose that redistribution of urea and creatinine, between serum and CSF, are useful biomarkers for evaluating disease-induced alterations in CSF biochemistry and BCSFB functional status.
Keywords: Cognitive impairment; Blood-brain barrier; CSF biomarkers; Choroid plexus
Introduction
Central nervous system (CNS) barrier leakiness in Alzheimer’s disease (AD) [1] involves injuries to choroid plexus (CP) [2-4] and the neurovascular unit (NVU) [5]. Intact barriers normally minimize blood constituent access to CNS [6]. Tight junction and barrier cell (endothelium and epithelium) restriction to diffusing solutes, along with active transport between CSF, brain and blood, create a stable, specialized neuronal interstitial environment and optimal CSF composition. Disruption of CP transporters and permeability, often attending disease-associated neurodegeneration, is reflected in CSF compositional changes.
Cognition is threatened [7] when damage to the blood-brain barrier (BBB) and blood-CSF barrier (BCSFB) destabilizes interstitial fluid in and CSF near, the hippocampus [8]. In this study, we emphasize permeability/transport changes at the BCSFB because our biochemical data were procured from samples of CSF, the fluid that originates mainly from CP tissues [2].
Albumin gauges substantially-increased pore size, e.g. as occurs in CNS barrier systems stressed with advanced neurodegeneration. The CSF-to-serum ratio (CSF/SER) for albumin has long been used to estimate BBB permeability, but strictly speaking, the CSF/SER value reflects primarily the altered structure/function at the BCSFB [9]. Upon gaining access to CSF, via CP, solutes of a wide range of molecular weights can impact cerebral homeostasis due to their accessibility to interstitial fluid across the permeable CSF-brain interfaces, e.g. ependyma.
Augmented CSF/SER values reflect greater permeation/retention of diffusing solutes in the CSF. The normally low CSF/SER of 0.005 for albumin (undergoing greatly restricted diffusion) increases in stroke, Parkinson’s disease dementia [10] and vascular cognitive impairment [11]. The large albumin molecule, however, may not sensitively detect diminutive-sized ‘openings’ in barriers presumably occurring at the earliest stages of MCI and AD.
Smaller biomarkers, like urea (mw 60), may probe more sensitively the associations between barrier disruptions and sequelae, e.g. impaired cognition. We propose a finely-discriminating marker, urea, with a mass ~1/1000 of albumin. Although previous clinical analyses have quantified CSF urea content, evidently earlier investigators did not propose urea as a potentially more sensitive marker of the CNS barrier leakiness that develops with progressing neurodegeneration. By detecting subtle changes in permeability, urea may allow earlier detection of CSF-brain fluid dys-homeostasis in MCI-AD. In humans, urea is subjected to molecular sieving by healthy barriers (CSF/SER of 0.75-0.8) [12]. We hypothesize that in MCI-AD, as barriers break down, urea diffusion is less restricted [13], causing CSF/SER to rise towards the equilibrium value of 1.0 for diffusing non-electrolytes such as urea.
Creatinine is another small biomarker candidate for CSF chemical (metabolite) analyses [14]. The normal value of 1.5-2.0 for CSF/ SER creatinine implicates active transport, potentially vulnerable in degenerating CP-barrier membranes. Our MCI-AD findings that steady-state CSF/SER creatinine substantially decreases, while CSF/ SER urea and albumin [15] increase, point to CNS barrier injury in cognitively-impaired patients. Such damage may include reduced CSF formation/flow, a factor affecting CSF solute (e.g. protein) concentrations [16]. This proposed new CSF small-biomarker approach to neuropathology/clinical analyses is potentially applicable to the early diagnosis and disease outcomes (e.g. therapeutic barrier tightening) from novel medicinals for MCI-AD.
Methods
Subjects and protocols
The Institutional Review Board of Rhode Island Hospital approved this investigation. It was a retrospective cross-sectional study of banked blood and CSF from 21 cognitively-impaired (CI) patients with either MCI (n=8) or AD (n=13). All patients were evaluated at the Rhode Island Hospital Alzheimer’s Disease and Memory Disorders Center over the period of 2010-2016. Over half the cohort was male (57%) in each subgroup (Table 1).
The main aim of our pilot study was to test for blood-CSF permeability differences in CI patients: MCI vs. AD. A secondary aim was to verify the expected differences between CI patients and healthy controls: 21 middle age adults with normal cognition and free of any known neuropathology or disruption of CNS barrier systems. Even though a rigorous ageindependent analysis was not feasible in our CI patients, because the mean age of the MCI subjects (69 years) was not significantly different from AD (68 years), we conducted a regression of all CSF/SER values against age; this included the 42 subjects in the 3 groups.
For controls, the inclusion criteria were patients who: 1) had diagnostic lumbar puncture at the Rhode Island, Miriam, or Newport Hospital Emergency Departments, 2) had a blood chemistry tube drawn as part of their diagnostic evaluation, 3) were adults 21 and older, and 4) provided specimens >0.5 ml for biochemical assays. Exclusion criteria were patients: 1) admitted to the hospital and under treatment (at the research time) that used their specimens, 2) whose CSF results were abnormal, including protein, glucose, Gram stain and cell counts, and 3) with a final discharge diagnosis of a central neurologic condition other than headache.
Eligible controls had an adequate volume of blood and CSF for analyses, and normal CSF protein, glucose, Gram stain, and cell counts. After collection, samples were aliquoted into 1 ml polypropylene tubes, capped/frozen at -80°C and scaled down for later analysis with commercial reagents by the Lifespan Core Research Laboratories. The majority of the samples from the 21 controls (Table 1) were obtained from cognitively-normal headache patients with an uneventful emergency room course and negative diagnostic work-up.
Markers selected for investigation
Molecules assessed were urea (MW 60), creatinine (MW 113) and albumin (MW ~68,000). All three are water-soluble with slow, gradual access to the CNS. Moreover, urea, creatinine and albumin in plasma (i.e., the source for brain uptake) originate mainly in the liver where they enter hepatic blood. Due to different sizes, these molecules are sterically hindered to various degrees (less restriction to lower molecular weight) when diffusing across barriers into CSF and brain. Urea usefully displays graded access by diffusion, not active transport [17], into the adult CNS. Urea is significantly smaller than the intermediate size (MW 300-500) water-soluble metabolites, endogenous compounds and drugs usually greatly restricted from entering the CNS. Analysis of small molecule penetration into CSF, to evaluate blood-CSF barrier status, was originally encouraged by Felgenhauer et al. [9].
Urea, creatinine and albumin concentrations were quantified in CSF and serum for absolute as well as relative concentrations (CSF/ SER partitioning) between CSF and plasma compartments. To enable functions in healthy CNS, such as CSF K homeostasis or CSF sink action on brain metabolites, various energized systems at the BCSFB normally maintain most solutes in CSF [18] at steady-state distributions (CSF/ SER) away from their equilibrium value of 1.
The working model
Overarching hypothesis: Progressive damage to brain barrier systems, as in MCI-AD, is reflected by changing values in the distribution of urea, creatinine and albumin, between plasma (serum) and CSF. Sub-hypothesis: The steady-state CSF/SER values of these substances in severe barrier breakdown (and thus loss of CSF sink action) will dissipate towards 1.0 expected for equilibrium distribution in a CNS energy-deprived state. With mitochondrial ATP depletion, as in neurodegeneration, the tight junctions and active transporters are hypofunctional, unable to maintain sufficient transbarrier gradients for solutes.
Statistical Analysis
Statistical parameters were obtained with Stata 14.1 (Stata Corp, College Station, TX).
Concentration ratios (CSF/SER), for paired CSF-serum samples, were determined for urea, creatinine and albumin. CSF/SER values were computed for CI patients (MCI-AD) vs. non-cognitively impaired controls. Data differences between groups were assessed with two-tailed Student’s t-test or the Mann-Whitney test. For aging analysis, a comparison of control vs. CI subjects was done by Pearson correlation coefficients, comparing age to BCSFB ratios, and to solute concentrations in serum and CSF. Statistical significance was defined as p<0.05.
Results
Demographic, diagnostic and psychometric findings
AD was the most common cognition-impairment diagnosis (62%, n=13), whereas the eight MCI patients comprised 38% of the cognitively-impaired subjects (Table 1). The Mini Mental State Examination mean score for the 21 patients was 23.5 (range 12-30). Gender representation was 43% female in the control and patient groups. We found significantly-decreased Aβ1-42/pTau in CSF in MCI and AD (Table 1).
Cerebrospinal fluid and serum composition
Urea, creatinine and albumin concentrations were quantified in 3 study groups (Table 1). Specimen data analyses revealed that the coefficients of variation (CV) for CSF urea of 23%, 24% and 19% (Controls, MCI and AD) and CSF creatinine (22%, 6.3% and 14.3%) were within or below the 20%-30% CV range typical for CSF analytes in quality-control measurements of CI-impaired patients.
Urea: Serum urea averaged 5.3 ng/μl in controls, increasing by ~35% to 7.0 and 6.9 ng/μl, respectively, in MCI (p<0.05) and AD (p<0.05). CSF urea was 4.1 ng/μl in healthy (non-CI) individuals, rising to 6.8 and 5.9 ng/μl in MCI (p<0.05) and AD (p<0.05). Thus CSF urea in cognitively-impaired patients was significantly elevated by ~50% (Table 1).
| Control N=21 |
MCI N=8 |
AD N=13 |
CI N=21 |
|
|---|---|---|---|---|
| Age | 45.6 (11.4) | 69.1 (7.2)* | 67.5 (11.3)* | 68.1 (9.7)* |
| Sex M/F | 12/9 | 7/1 | 5/8 | 12/9 |
| MMSE | ND | 26.4 (3.1) | 21.7 (5.8) | 23.5 (5.4) |
| Urea, serum | 5.29 (1.47) | 7.04 (1.74)* | 6.86 (1.04)* | 6.93 (1.31)* |
| Urea, CSF | 4.12 (0.94) | 6.79 (1.62)* | 5.91 (1.15)* | 6.24 (1.38)* |
| Urea CSF/serum ratio | 0.80 (0.15) | 0.98 (0.17)* | 0.86 (0.08) | 0.91 (0.14)* |
| Creatinine, serum | 4.11 (0.85) | 4.23 (0.79) | 4.00 (0.52) | 4.09 (0.63) |
| Creatinine, CSF | 6.13 (1.34) | 4.64 (0.29)* | 4.52 (0.65)* | 4.57 (0.53)* |
| Creatinine CSF/serum ratio | 1.52 (0.36) | 1.13 (0.20)* | 1.14 (0.13)* | 1.13 (0.16)* |
| Albumin, serum | 92.81(29.72) | 106.83(27.51)* | 112.37(20.44)* | 106.42 (27.5)* |
| Albumin, CSF | 0.402 (0.196) | 0.530 (0.163) |
0.713* (0.297) |
0.655 (0.271) |
| Albumin CSF/serum ratio | 0.0045 (0.0023) | 0.0051 (0.0030) |
0.0065 (0.0031) | 0.0064 (0.003) |
| CSF Aß1-42/pTau | 8.2 (1.4) | 5.8 (2.3) * | 6.0 (1.0) * | 5.9 (1.3) * |
All continuous measures are presented as mean+standard deviation
MMSE was not determined for cognitively-normal control subjects
MCI: Mild Cognitive Impairment; AD: Alzheimer’s Disease; CI: Cognitive Impairment (pooled data from MCI and AD patients)
Concentrations of urea and creatinine are in ng/µl; albumin in µg/µl
*p<0.05, MCI or AD or CI vs. Control
Table 1: Demographic, psychometric and biochemical data for the study of CSF and serum composition in patients with cognitive impairment.
Creatinine: The serum creatinine concentration of 4.1 ng/μl in controls was not significantly altered (p>0.05) in MCI (4.2 ng/μl) or AD (4.0). This indicated stable kidney function in the 3 groups. On the other hand, compared to controls, creatinine in CSF decreased by ~25% in both MCI and AD (p<0.05). Our control of 54+12 (SD) μM for CSF creatinine is similar to the 68+18 μM in another human CSF study [19].
Albumin: Serum albumin averaged 92.8 μg/μl in controls, increasing in MCI to 107 (p<0.05) and 112 in AD (p<0.05) (Table 1). Correspondingly, in CSF, albumin was 0.402 μg/μl in controls, increasing to 0.530 and 0.710 μg/μl, respectively, in MCI and AD (p<0.05).
Concentration ratios: Cerebrospinal fluid to serum
Steady-state concentration ratios inform on the degree and direction (vector) of solute partitioning. CSF/SER values 1.0 usually indicates active transport from plasma into CNS. CSF/ SER urea was normally (controls) ~0.80 but increased over the range of 0.86 to 0.98 (p<0.05) in cognitively-impaired (CI) individuals (Table 1). Mean CSF/SER for creatinine was ~1.52 in controls, significantly decreasing to ~1.14 in MCI-AD (p<0.05) (Table 1). CSF/SER albumin tended to increase in MCI-AD (p=0.08) (Table 1). CSF/SER 95% confidence intervals in controls and CI were: urea (0.68, 0.80) and (0.80, 0.92); creatinine (1.3, 1.6) and (0.97, 1.1); albumin (0.004, 0.006) and (0.004, 0.015).
CSF/SER data for MCI and AD were indistinguishable (p>0.05). Therefore we graphed (box-and-whisker) pooled data for all the cognitively-impaired (CI) patients. The generated median, together with the 25th and 75th percentiles are presented for each solute (Figure 1).
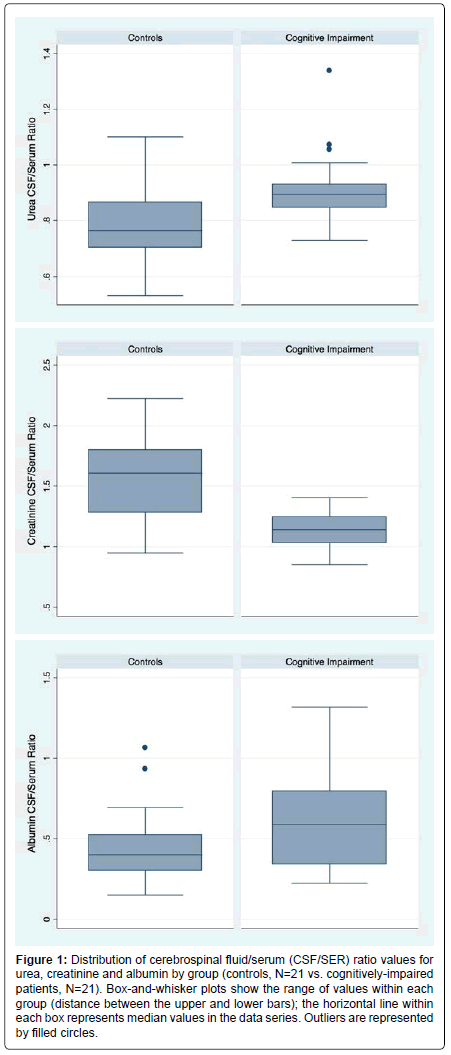
Figure 1:Distribution of cerebrospinal fluid/serum (CSF/SER) ratio values for urea, creatinine and albumin by group (controls, N=21 vs. cognitively-impaired patients, N=21). Box-and-whisker plots show the range of values within each group (distance between the upper and lower bars); the horizontal line within each box represents median values in the data series. Outliers are represented by filled circles.
Pearson correlation coefficients comparing age to BCSFB ratios (CSF/SER) revealed no significant relationship between age and ratios for urea, creatinine or albumin among the subgroups of normal and cognitively-impaired (CI) subjects (Table 2). The same held true for urea for the total group of subjects. Significant relationships between age and the ratio for creatinine (r=-0.42; p=0.006) as well as the ratio for albumin (r=0.32 and p=0.04) were seen for the group as a whole, though the significance level for albumin would be non-significant, i.e., p>0.05 if corrected for multiple comparisons.
| Control Subjects N=21 |
Cognitively Impaired Subjects (AD+MCI) N=21 |
All Subjects N=42 |
||||
|---|---|---|---|---|---|---|
| r | p | r | p | r | p | |
| Urea, serum | 0.06 | 0.79 | 0.51 | 0.02 | 0.53 | 0.0003 |
| Urea, CSF | 0.04 | 0.85 | 0.40 | 0.07 | 0.61 | <0.0005 |
| Urea CSF/serum ratio | -0.21 | 0.61 | -0.08 | 0.74 | 0.20 | 0.20 |
| Creatinine, serum | 0.01 | 0.97 | 0.41 | 0.07 | 0.10 | 0.53 |
| Creatinine, CSF | 0.10 | 0.67 | 0.29 | 0.20 | 0.38 | 0.01 |
| Creatinine CSF/serum ratio | 0.10 | 0.68 | -0.18 | 0.44 | -0.42 | 0.006 |
| Albumin, serum | 0.38 | 0.08 | 0.01 | 0.99 | 0.39 | 0.02 |
| Albumin, CSF | 0.46 | 0.04 | 0.31 | 0.18 | 0.44 | 0.004 |
| Albumin CSF/serum ratio | 0.14 | 0.55 | 0.29 | 0.22 | 0.32 | 0.04 |
Table 2: Comparison of control vs. CI subjects by Pearson correlation coefficients comparing age to BCSFB ratios and to solute concentrations in serum and CSF.
Discussion
The key finding for cognitively-impaired patients is the dissipated molecular gradients of certain solutes between CSF and serum. Thus in MCI-AD patients, the mean CSF/SER values for urea, creatinine and albumin all became closer to 1.0. This results from BCSFB breakdown. Cognition depends upon barrier integrity, essential for optimal neurotransmission and mentation [18]. Barrier breaching, by disturbing CSF-cerebral homeostasis, leads to cognitive impairment.
A discussion of CSF/SER places in perspective the current ratio analyses. Typical CSF/SER values in health are 0.75 for potassium [18], 0.03 for iodide, 1.1 for chloride [12], 1.3 for magnesium, ~4 for folate and ascorbate [20] and ~0.005 for several proteins originating from plasma [18].
The control CSF/SER values of 0.80 for urea, 1.5 for creatinine and 0.0045 for albumin were significantly changed in MCI and AD (Table 1), reflecting altered BCSFB characteristics and modified CSF biochemistry.
CSF composition changed as much in MCI as AD. Our baseline (control) data agrees with previous CSF/SER determinations in healthy humans and animals [18,21,22]. The lack of statistical difference between MCI and AD may relate to a plateau effect for urea and creatinine, i.e., at the stage of MCI analyzed, the barrier problem had already become maximal. Given similar ages of the MCI/AD patients (68-69 years), the age factor does not explain the barrier/compositional disruptions in these two CI groups. By Pearson correlation analysis, there is no evidence (Table 2) that age confounded the interpretation of altered values in CI patients. Below we discuss factors determining CSF/ SER and how these parameters can usefully biomark pathophysiology.
Permeability considerations for the BCSFB
Urea ‘detects’ tiny openings in barriers. Neither actively transported at the BCSFB/BBB [22] nor bound to plasma proteins [21], urea gradually diffuses into CSF and brain. Tracer kinetics reveal slow 14C-urea permeation from plasma into CSF-CNS [23] (~8 h to steady state), the slowness caused by the diffusion-impeding tight junctions between sound barrier cells [13]. Choroidal tight junctions, damaged by β-amyloid in neurodegenerative diseases [24], become more permeable as paracellular pore size widens [13]. Matrix metalloproteinases, activated by β-amyloid in AD models [25], contribute to tight junction destruction [26]. Reduced CP claudin-5 protein expression in AD [27] also promotes tight junction leakiness [8,27]. This enhances penetration of urea (and albumin) between epithelial cells of BCSFB [3], resulting in increased CSF and brain interstitial fluid concentrations of these plasma solutes [28].
Kaiser et al. also noted elevated urea in AD CSF [29]. This fits our observation that CSF/SER urea increased to 0.90-0.95 in MCI-AD. The range of values likely relates to urea penetration being proportional to tight junction injury [13]. Excessive urea in CSF in MCI-AD results from barrier breaching, plus the greater plasma urea concentration driving the diffusion. (If BCSFB remained intact, the CSF/SER of diffusing solutes would be unaffected by rising plasma concentration.) BCSFB remains intact to 14C-urea in uremia [30], intimating that elevated plasma urea per se in MCI-AD does not modify choroidal permeability. Information is needed on how the ~50% increase in CSF urea in CI affects cerebral functions.
Analysis of macromolecular albumin (Table 1), the longstanding ‘permeability standard’, verifies AD findings of BCSFB breakdown. CSF albumin rises due to: i) greater flux across compromised BCSFB, and ii) reduced CSF sink action as fluid formation decreases in neurodegeneration. CSF flow reduction augments protein retention [16]. Lower CSF flow and pulsation, in AD patients [31] and old animals [32], contributes to elevated CSF/SER for albumin in MCI-AD, especially when CSF sampling is from the lumbar region [33]. This CSF flow-protein concentration inverse relationship increases the effective permeability of BCSFB, so that in AD more protein stays longer in CSFCNS. Systematic MRI studies of CSF dynamics in MCI are needed to evaluate CSF protein buildup in relation to failing cognition.
Transporter considerations for the BCSFB
Transport activity by CP greatly impacts CSF composition [6]. CSF creatinine is complex to interpret because, in addition to BCSFB transport activity, there are brain metabolic factors as well. Human CSF concentrates creatinine to a level of ~1.5 (present study) to 2.1 times [19] that in serum. Evidence is lacking for active transport of plasma creatinine into CSF or brain. Moreover, Bradbury et al. found highlyrestricted 14C-creatinine diffusion from plasma into CNS [22]. How to explain, then, the normal CSF/SER creatinine accumulation of 1.5- fold? Brain formation of creatinine from creatine [14], transported into CNS [34,35], evidently builds up CSF creatinine to a level ~50% above plasma; any CSF excess of creatinine is actively reabsorbed by CP into blood [14]. Homeostasis of CSF creatinine minimizes seizures.
Our finding of a 25% diminished CSF creatinine in MCI-AD raises the issue about the optimal level for brain function. Does this diminution imply less creatine-phosphate energy availability for CNS [36] that would generate less metabolite end-product creatinine? Potential explanations for dissipated CSF/SER creatinine in cognitivelyimpaired patients are: i) less active transport of creatine across an altered BBB into brain (therefore, less creatinine generated from lower brain creatine concentration), ii) a creatinine diffusion leak from brain and CSF into blood, down a gradient across more permeable transport interfaces, and iii) stimulation of the actively-transporting OCT3 in CP that removes creatinine from CSF [14]. These explanations are consistent with the lower CSF creatinine in MCI-AD.
Creatine-creatinine interactive dynamics in brain energy metabolism is impacted by activity of the CP and BBB transporters (for creatine and glucose delivery, also decreased in AD [37]) and by the adverse interactions of β-amyloid with creatine (and its kinase) to form creatine deposits [38,39]. Therefore less creatine (and glucose [37]) availability to neuronal mitochondria, due to altered BBB transport [36] or β-amyloid toxicity, would lower central energy production and impair metabolism. Creatinine as a CSF biomarker, to track brain creatine metabolism (indirectly by putative creatinine diffusion from brain interstitium to CSF [9]), may allow earlier detection of MCI and possible treatment with creatine [38] to curtail AD complications.
Clinical implications of findings
CSF composition is a critically-important assessment clinically. This is true because solutes in CSF, by flow dynamics, have access to neuronal networks brain-wide. CSF and its constituents penetrate CNS: i) from the ventricles, by highly-permeable transependymal movement into interstitium [28], and ii) from the cortical subarachnoid space, into the periarterial flow pathway [40], i.e., the proximal part of the glymphatic system. Stability of CSF and homeostasis of the serially-proximate extracellular fluid are intimately linked. Multiple CSF compositional changes, away from the normal steady-state distribution, contribute to cognitive impairment because CSF neurochemical changes (dyshomeostasis) are rapidly/extensively transmitted to the brain interior. Neurons, exquisitely sensitive to biochemical alterations in extracellular fluid, vitally depend on the purity/stability of CSF as maintained by CP.
We did not anticipate that CSF/SER for urea (and albumin) would increase more in some MCI patients than AD. Consistently, though, the biomarker index severity for CSF Aβ1-42/pTau was most prominent in MCI, again pointing to barrier/CSF distortions occurring relatively early in progressive cognition loss. Interestingly, two CNS barrier-injury molecules, the high-mobility group box protein 1 and thrombomodulin, have a greater concentration in serum in MCI [41] than in AD and controls. Their elevated titers in MCI may partly explain the induced experimental increase in CNS barrier paracellular permeability [41] and neuroinflammation ensuing from serum leakage.
Our cohort data are not extensive enough to determine if APOE genotype explains the substantially-increased permeability in MCI. APOE4 is a risk factor for BBB damage [42,43] and MCI onset [44]. Possibly more people in our MCI group (vs. AD) may be homo- or heterozygous for APOE4. APOE4 has been linked to disordered lipid metabolism [45]. In CP, the 4 allele interferes with cholesterol release into CSF [46]; consequently, accumulated cholesterol in the choroidal epithelium may interfere with [46] transport and CSF formation rate, thereby disrupting BCSFB integrity. Larger cohorts of MCI-AD can reveal how APOE4 status affects urea/protein concentration in CSF, as well as the creatine-creatinine system.
The currently-described barrier/neurochemical profiling of CSF/ SER for urea and creatinine opens the door for new vistas in assessing relationships among BCSFB function, CSF biochemistry and cognitive status. Expanded studies of small molecule CSF biomarkers [47] will help fill a gap in the wide spectrum of transport/metabolic abnormalities in MCI vs. AD.
Conclusion
This study demonstrates that BCSFB (choroid plexus) function is altered in MCI, causing redistribution of two small non-electrolytes, urea and creatinine, between serum and CSF. Unexpectedly, the altered CSF/serum ratios and distorted CSF composition were as great in MCI as in AD. Further studies, on longitudinal aspects of neurodegeneration progression, can test the postulate that BCSFB ‘opening’ in MCI precedes barrier breaching in AD. APOE4 analyses have historically dealt mainly on BBB permeability; however, increasing attention on β4 allele actions on BCSFB permeability/CSF composition in MCI-AD will provide a more comprehensive understanding of the CSF-brain pathophysiologic nexus. Monitoring the CSF urea, creatinine and other small molecules, in both pre-MCI and CI subjects, affords biomarking opportunities to evaluate early, possibly differential metabolic distortions relating to BCSFB functional status in patients approaching/experiencing neurodegeneration.
Acknowledgment
This study was supported by institutional funds provided to Dr. Ott and Stopa from Rhode Island Hospital. The authors thank Lifespan IRB reviewers at Rhode Island Hospital for evaluating the study when proposed. Professor K Hosoya (University of Toyama) kindly provided helpful comments. We thank Charles Denby, manager of the specimen bank in Neurology (Rhode Island Hospital), for handling/delivering CSF and serum samples for analysis.
References
- Bowman GL, Kaye JA, Moore M, Waichunas D, Carlson NE, et al. (2007) Blood-brain barrier impairment in Alzheimer disease: Stability and functional significance. Neurology 68: 1809-1814.
- Spector R, Keep RF, Snodgrass SR, Smith QR, Johanson CE (2015) A balanced view of choroid plexus structure and function: Focus on adult humans. Exp Neurol 267: 78-86.
- Johanson C, McMillan P, Tavares R, Spangenberger A, Duncan J, et al. (2004) Homeostatic capabilities of the choroid plexus epithelium in Alzheimer's disease. Cerebrospinal Fluid Res 1: 3.
- Johanson CE (2017) Choroid plexus-cerebrospinal fluid transport dynamics: Support of brain health and a role in neurotherapeutics: Elsevier Inc., Academic Press.
- Sweeney MD, Sagare AP, Zlokovic BV (2015) Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer's disease. J Cereb Blood Flow Metab 35: 1055-1068.
- Johanson CE, Duncan JA, Klinge PM, Brinker T, Stopa EG, et al. (2008) Multiplicity of cerebrospinal fluid functions: New challenges in health and disease. Cerebrospinal Fluid Res 5: 10.
- Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, et al. (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85: 296-302.
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA (2007) Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab 27: 697-709.
- Felgenhauer K, Liappis N, Nekic M (1982) Low molecular solutes and the blood cerebrospinal fluid barrier. Klin Wochenschr 60: 1385-1392.
- Llorens F, Schmitz M, Gloeckner SF, Kaerst L, Hermann P, et al. (2015) Increased albumin CSF/serum ratio in dementia with Lewy bodies. J Neurol Sci 358: 398-403.
- Candelario-Jalil E, Thompson J, Taheri S, Grossetete M, Adair JC, et al. (2011) Matrix metalloproteinases are associated with increased blood-brain barrier opening in vascular cognitive impairment. Stroke 42: 1345-1350.
- Sambrook MA (1974) The relationship between cerebrospinal fluid and plasma electrolytes in patients with meningitis. J Neurol Sci 23: 265-273.
- Murphy VA, Johanson CE (1985) Adrenergic-induced enhancement of brain barrier system permeability to small nonelectrolytes: Choroid plexus versus cerebral capillaries. J Cereb Blood Flow Metab 5: 401-412.
- Tachikawa M, Kasai Y, Takahashi M, Fujinawa J, Kitaichi K, et al. (2008) The blood-cerebrospinal fluid barrier is a major pathway of cerebral creatinine clearance: Involvement of transporter-mediated process. J Neurochem 107: 432-442.
- Ott BR, Cohen RA, Gongvatana A, Okonkwo OC, Johanson CE, et al. (2010) Brain ventricular volume and cerebrospinal fluid biomarkers of Alzheimer's disease. J Alzheimers Dis 20: 647-657.
- Reiber H (1994) Flow rate of cerebrospinal fluid (CSF)--a concept common to normal blood-CSF barrier function and to dysfunction in neurological diseases. J Neurol Sci 122: 189-203.
- Berger UV, Tsukaguchi H, Hediger MA (1998) Distribution of mRNA for the facilitated urea transporter UT3 in the rat nervous system. Anat Embryol (Berl) 197: 405-414.
- Davson H, Welch K, Segal M (1987) The physiology and pathophysiology of the cerebrospinal Fluid. New York: Churchill Livingstone.
- Deignan JL, De Deyn PP, Cederbaum SD, Fuchshuber A, Roth B, et al. (2010) Guanidino compound levels in blood, cerebrospinal fluid and post-mortem brain material of patients with argininemia. Mol Genet Metab 100: S31-S36.
- Spector R, Johanson CE (2014) The nexus of vitamin homeostasis and DNA synthesis and modification in mammalian brain. Mol Brain 7: 3.
- Johanson CE, Woodbury DM (1978) Uptake of [14C]urea by the in vivo choroid plexus--cerebrospinal fluid--brain system: Identification of sites of molecular sieving. J Physiol 275: 167-176.
- Bradbury MW, Davson H (1964) The transport of urea, creatinine and certain monosaccharides between blood and fluid perfusing the cerebral ventricular system of rabbits. J Physiol 170: 195-211.
- Parandoosh Z, Johanson CE (1982) Ontogeny of blood-brain barrier permeability to and cerebrospinal fluid sink action on [14C]urea. Am J Physiol 243: R400-R407.
- Gonzalez-Marrero I, Gimenez-Llort L, Carmona-Calero E, Castaneyra-Ruiz L, Brito-Armas JM, et al. (2015) Choroid plexus dysfunction in a triple transgenic mouse model of Alzheimer’s disease. Front Cell Neurosci 9: 17.
- Brkic M, Balusu S, Van Wonterghem E, Gorlé N, Benilova I, et al. (2015) Amyloid ß oligomers disrupt blood-CSF barrier integrity by activating matrix metalloproteinases. J Neurosci 35:12766-12778.
- Steeland S, Gorlé N, Vandendriessche C (2018) Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer's disease. EMBO Mol Med.
- Bergen AA, Kaing S, ten Brink JB, Gorgels TG, Janssen SF, et al. (2015) Gene expression and functional annotation of human choroid plexus epithelium failure in Alzheimer's disease. BMC Genomics 16: 956.
- Spector R, Robert Snodgrass S, Johanson CE (2015) A balanced view of the cerebrospinal fluid composition and functions: Focus on adult humans. Exp Neurol 273: 57-68.
- Kaiser E, Schoenknecht P, Kassner S, Hildebrandt W, Kinscherf R, et al. (2010) Cerebrospinal fluid concentrations of functionally important amino acids and metabolic compounds in patients with mild cognitive impairment and Alzheimer's disease. Neurodegener Dis 7: 251-259.
- Hise MA, Johanson CE (1979) The sink action of cerebrospinal fluid in uremia. Eur Neurol 18: 328-337.
- Silverberg GD, Heit G, Huhn S, Jaffe RA, Chang SD, et al. (2001) The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer's type. Neurology 57: 1763-1766.
- Preston JE (2001) Ageing choroid plexus-cerebrospinal fluid system. Microsc Res Tech 52: 31-37.
- Asgari M, de Zélicourt DA, Kurtcuoglu V (2017) Barrier dysfunction or drainage reduction: Differentiating causes of CSF protein increase. Fluids Barriers CNS 14: 14.
- Ohtsuki S, Tachikawa M, Takanaga H, Shimizu H, Watanabe M, et al. (2002) The blood-brain barrier creatine transporter is a major pathway for supplying creatine to the brain. J Cereb Blood Flow Metab 22: 1327-1335.
- Hosoya K, Tachikawa M (2011) Roles of organic anion/cation transporters at the blood-brain and blood-cerebrospinal fluid barriers involving uremic toxins. Clin Exp Nephrol 15: 478-485.
- Uemura T, Ito S, Ohta Y, Tachikawa M, Wada T, et al. (2017) Abnormal N-Glycosylation of a novel missense creatine transporter mutant, G561R, associated with cerebral creatine deficiency syndromes alters transporter activity and localization. Biol Pharm Bull 40: 49-55.
- Harik SI, Kalaria RN (1991) Blood-brain barrier abnormalities in Alzheimer's disease. Ann N Y Acad Sci 640: 47-52.
- Burklen TS, Schlattner U, Homayouni R, Gough K, Rak M, et al. (2006) The creatine kinase/creatine connection to Alzheimer's disease: CK-inactivation, APP-CK complexes and focal creatine deposits. J Biomed Biotechnol 2006: 35936.
- Gallant M, Rak M, Szeghalmi A, Del Bigio MR, Westaway D, et al. (2006) Focally elevated creatine detected in amyloid precursor protein (APP) transgenic mice and Alzheimer disease brain tissue. J Biol Chem 281: 5-8.
- Plog BA, Nedergaard M (2018) The glymphatic system in central nervous system health and disease: past, present and future. Annu Rev Pathol 13: 379-394.
- Festoff BW, Sajja RK, van Dreden P, Cucullo L (2016) HMGB1 and thrombin mediate the blood-brain barrier dysfunction acting as biomarkers of neuroinflammation and progression to neurodegeneration in Alzheimer's disease. J Neuroinflammation 13: 194.
- Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, et al. (2007) Microvascular injury and blood-brain barrier leakage in Alzheimer's disease. Neurobiol Aging 28: 977-986.
- Donahue JE, Johanson CE (2008) Apolipoprotein E, amyloid-beta and blood-brain barrier permeability in Alzheimer disease. J Neuropathol Exp Neurol 67: 261-270.
- Scarabino D, Broggio E, Gambina G, Maida C, Gaudio MR, et al. (2016) Apolipoprotein E genotypes and plasma levels in mild cognitive impairment conversion to Alzheimer's disease: A follow-up study. Am J Med Genet B Neuropsychiatr Genet 171: 1131-1138.
- Poirier J, Miron J, Picard C, Gormley P, Théroux L, et al. (2014) Apolipoprotein E and lipid homeostasis in the etiology and treatment of sporadic Alzheimer's disease. Neurobiol Aging 35: S3-S10.
- Fujiyoshi M, Ohtsuki S, Hori S, Tachikawa M, Terasaki T (2007) 24S-hydroxycholesterol induces cholesterol release from choroid plexus epithelial cells in an apical- and apoE isoform-dependent manner concomitantly with the induction of ABCA1 and ABCG1 expression. J Neurochem 100: 968-978.
- Heister D, Brewer JB, Magda S, Blennow K, McEvoy LK (2011) Predicting MCI outcome with clinically available MRI and CSF biomarkers. Neurology 77: 1619-1628.
Citation: Johanson CE, Stopa EG, Daiello L, de la Monte S, Keane M, et al. (2018) Disrupted Blood-CSF Barrier to Urea and Creatinine in Mild Cognitive Impairment and Alzheimer’s Disease. J Alzheimers Dis Parkinsonism 8: 435. DOI: 10.4172/2161-0460.1000435
Copyright: ©2018 Johanson CE, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Select your language of interest to view the total content in your interested language
Share This Article
Recommended Journals
Open Access Journals
Article Tools
Article Usage
- Total views: 9476
- [From(publication date): 0-2018 - Dec 21, 2025]
- Breakdown by view type
- HTML page views: 8374
- PDF downloads: 1102