Research Article Open Access
PCR-Free Ultrasensitive Capacitive Biosensor for Selective Detection and Quantification of Enterobacteriacea DNA
Ally Mahadhy1, Gashaw Mamo1, Eva Ståhl-Wernersson1, Bo Mattiasson1,2 and Martin Hedström1,2*
1Department of Biotechnology, Lund University, P.O. Box 124, SE-22100 Lund, Sweden
2CapSenze HB, Medicon Village, SE-22838 Lund, Sweden
- *Corresponding Author:
- Martin Hedström
Department of Biotechnology,Lund University
P.O. Box 124, SE-22100 Lund, Sweden
Tel: +46 46-222 75 78
Fax: +46 46-222 47 13
E-mail: Martin.Hedstrom@biotek.lu.se
Received date: September 12, 2014; Accepted date: October 07 2014; Published date: October 10, 2014
Citation: Mahadhy A, Mamo G, Ståhl-Wernersson E, Mattiasson B, Hedström M (2014) PCR-Free Ultrasensitive Capacitive Biosensor for Selective Detection and Quantification of Enterobacteriacea DNA. J Anal Bioanal Tech 5:210 doi: 10.4172/2155-9872.1000210
Copyright: © 2014 Mahadhy A, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Analytical & Bioanalytical Techniques
Abstract
This paper presents a flow-based ultrasensitive capacitive biosensor for the detection of bacterial DNA. The used sensor chip consists of a gold electrode, insulated with a polytyramine layer and covalently tagged with a DNA capture probe. The hybridization of target DNA to the capture probe resulted in sensor response. The sensor response was linear vs. log concentration in the range 1.0 × 10-12 to 1.0 × 10-7 moles per litre with a detection limit of 6.5 × 10-13 M. An alternative approach to bacterial DNA sample preparation for a flow-based analysis is also reported. The approach involved application of a thermostable ssDNA binding protein to prevent re-annealing of a heatdenatured target DNA prior to analysis. During analysis, formamide was integrated in the running buffer to denature ET SSB. E. coli DNA corresponding to 10 cells per millilitre of sample was detected in 15 min by this DNA-sensor. The sensor chip could be re-used up to 20 times with RSD of < 6%. The DNA-sensor chip was able to discriminate between Enterobacteriaceae (E. coli) and Lactobacillaceae (L. reuteri) DNAs. The reported DNA-sensor lays the groundwork for incorporating the method into an integrated system for in-field bacteria detection.
Keywords
DNA-sensor; Capacitance; Formamide; Enterobacteriaceae; ssDNA-binding-protein
Introduction
There is a need for rapid and field-adopted assays for detection of pathogenic microorganisms. This need is reflected in most diagnostic assays of pathogenic organisms that are still made in clinical laboratories. For instance, the source of food-borne illness is determined in less than 50% of the cases, usually because of limited diagnostic capabilities [1,2]. Most of the hospital, clinical and quality control laboratories use a traditional method, plate counting, as a primary method for detection and quantification of infectious pathogens. The method is laborious and time consuming; it takes 24 to 48 hr to obtain initial results [3,4].
The limitations of diagnostic capabilities have greatly increased the demand for developing a fast, selective and sensitive bioanalytical technique for the detection and quantification of infectious pathogens in the samples. Various DNA-based sensor models have been reported for detection of different pathogenic organisms; E. coli [5-7]; Neisseria meningitidis [8; Staphylococcus aureus [9]; and Mycobacterium tuberculosis [10]. The DNA-sensors make use of genetic markers of microorganisms for their identification and quantification. A genetic marker could be a DNA sequence that is responsible for pathogenicity or a unique DNA sequence for a particular microorganism [11]. However, a main problem in detection of DNA at physiological levels is that the target DNA is in a very low concentration, usually lower than the detection limits of many reported analytical techniques. Thus, amplification of the sample by polymerase chain reaction (PCR) has been the best choice [12]. On the other hand, the use of PCR is not only expensive but it is also limited in part by presence of inhibitory substances in complex biological samples, which reduce or even block the amplification capacity of PCR. For example, PCR mixture containing widely used Taq polymerase is totally inhibited by the presence of 0.004% (v/v) red blood cells [13].
This paper presents a real-time, PCR-free, selective and sensitive capacitive DNA-sensor, for the identification and quantification of Enterobacteriaceae DNA. Rod-shaped bacteria of the Enterobacteriacea family are one of the major worldwide health hazards and particularly in developing countries where sanitation standards are low. Strains of Escherichia coli, Shigella, Salmonella and Yersinia pestis are responsible for diarrhea, severe bacillary dysentery, typhoid and other intestinal diseases, as well as genitourinary tract and blood infections. According to the World Health Organization report, there are 4.5 billion cases every year, out of which approximately 1.9 million have a lethal outcome. This makes intestinal infections the third most mortal group of human diseases [14].
We have developed a DNA based sensor, as DNA probes are more cheaper and reliable than protein probes. As such, antibodies are very expensive and the proteins that they target can be degraded during sample preparation, where as targeted ssDNAs by ssDNA probes are much more stable.
The hybridization between the capture DNA probes and targeted DNAs was monitored by capacitance measurements [15]. E. coli BL21 (DE3), family Enterobacteriaceae was selected as a candidate and model for this study. However, the capacitive DNA-sensor can be used to target a broader range of bacteria that share a certain gene. Time consumption and sensitivity of the capacitive DNA-sensor and the total plate counting technique were compared as well.
Experimental section
Reagents
Single stranded 30-mer oligonucleotides; capture probe and target (complementary) probe designed from E. coli BL21(DE3) 16S rDNA were custom-made by Intergrated DNA Technologies, Inc. (Leuven, Belgium), while 16S rDNA universal primers: forward (63F) and reverse (1389R) were available in our lab. The sequences of all oligonucleotides are listed in Table 1. Extremely thermostable single-stranded DNA binding protein (ET SSB) was purchased from New England Biolabs (Ipswich, MA, USA). Restriction enzyme, Fastdigest AciI (SsiI) from Fermentas (Glen Burnie, MD, USA). Dream Taq PCR master mix (2X); GeneRuler Ultra low range DNA ladder and GeneRuler (1 kb) were all purchased from Thermo Fisher Scientific, Inc. (Göteborg, Sweden). DNA extraction/purification kit (ZR Fungal/Bacterial DNA MiniPrepTM) was purchased from Zymo research corporation (Irvine, CA, USA), and 5X Phusion buffer from Finnzymes (Espoo, Finland). Ethanol, sodium hydroxide and Omega Bio-tek’s gel extraction kit were obtained from VWR International (Leuven, Belgium). Tyramine, N-hydroxysuccinimide (NHS), N-(3- dimethylaminopropyl) N-ethylcarbodiimide hydrochloride (EDC), agarose type I, Luria-Bertani (LB) agar, formamide and 1-dodecane thiol were obtained from Sigma-Aldrich (Steinheim, Germany). E. coli BL21 (DE3) strain; L. reuteri DSM 20016; were available in our Department. All other chemicals used were of analytical grade. Buffers and solutions were prepared with double distilled water from a Milli-Q system by Millipore (Massachusetts, USA). Buffers were filtered through nitrocellulose membrane of a pore size 0.22 μm, from Sigma- Aldrich (Steinheim, Germany), and degassed prior to use.
Instruments
NanoDrop ND 1000 UV-Vis spectrophotometer, from NanoDrop Technologies (San Jose, CA, USA), an ultrasonicator, Hielscher UP 400S (Teltow, Germany), a water bath from Haake TP 42 (Karlsruhe, Germany), a cryostat, Neslab RTE-200 (Portsmouth, NH, USA), a PCR machine, Thermocycler Biometra RS 232 (Gottingen, Germany). Light-inverted microscope from Nikon, (Japan), Gel electrophoresis equipment; Electrophoresis Power Supply/ EPS 500/400 was from Pharmacia Fine Chemicals, Inc. (New Jersey, USA), while a gel visualizing equipment, MiniBis Pro 16 mm was from DNR Bio- Imaging system Ltd (Jerusalem, Israel). Plasma cleaning system from Harrick (New York, USA), a column thermostat (oven), Jetstream 2 from LabVision (Vienna, Austria) and Fast data acquisition unit from Keithley Instruments (Clevland, OH, USA).
Procedures
Preparation of working electrode: The study used a custommade 3 mm diameter disposable gold working electrode (Academic workshop, Linköping University, Sweden). The electrode was initially cleaned in acetone, then followed by ethanol and finally piranha solution (3:1) concentrated sulfuric acid: 30% hydrogen peroxide) for 5 min in each solution, while sequentially rinsed with distilled water before being placed in the plasma cleaning for 20 min. The cleaned working electrode was coated by electropolymerization of tyramine on the electrode surface [16]. The coated electrode was then rinsed with distilled water to remove any loosely bound polymer. The electrode was carefully dried with a stream of nitrogen gas after each step.
The immobilization of the capture probe on the surface of polytyramine coated electrode was carried out under optimized conditions [17], by applying 20 μL of 10 μM capture probe (DNA) in 10 mM potassium phosphate buffer (PB) solution, pH 7.2, containing 5 mM EDC, 8 mM NHS. The electrode was incubated during two hours at room temperature. After immobilization, DNA modified electrode was rinsed with double distilled water and 10 mM PB, pH 7.2, with 30% formamide. Finally, the electrode was immersed in 1-dodecanethiol (10 mM in ethanol) in order to block pinholes in the affinity surface [15], and was then stored at 4°C until use.
Capacitance measurements: The prepared working electrode was mounted in a four-electrode flow-injection cell with a dead volume of 10 μL and connected to a potentiostat, two platinum wires; one was used as the auxilary and the other as the reference electrode. An external reference (Ag/AgCl) was placed in the outlet stream. A fast data acquisition unit was connected between a personal computer and the potentiostat. To reduce the noise in the system, the potentiostat is powered from data acquisition unit, which in turn was powered by the computer through a galvanometrically insulated power supply. A schematic drawing of the system set up is provided elsewhere [15], the four-electrode flow-injection cell with the mounted electrodes was kept in the column thermostat; the working (hybridization) temperature was kept at 30°C, unless stated otherwise. The mobile phase (10 mM PB, pH 7.2, with 30% formamide) was infused using a peristaltic pump at a flow rate of 100 μL min-1. Samples were injected into the flow via a 250 μL loop. The DNA-sensor was regenerated with 0.5 M NaOH after each sample injection. The total capacitances and capacitance change (ΔC) were calculated from the current response as previously described in details [15]. All experiments were performed in triplicates.
Preparation of calibration curve: A standard stock solution of 1.0 × 10-5 M synthesised 30-mer ss16S rDNA, which is complementary to the capture probe, was prepared and serially diluted by a factor of 10, to make 9 solutions of concentrations from 1.0 × 10-6 to 1.0 × 10-14 M. Aliquots of each concentration and a blank (mobile phase) were injected in triplicate and capacitance change (ΔC) for each sample was calculated. A calibration curve of a logarithmic concentration of injected DNA sample versus registered signal was plotted. From the calibration curve, a linear range and the limit of detection (LOD) was determined. Using the same plotted curve as a reference, DNA samples from bacteria were analysed.
Re-usability: The re-usability of 30-mer 16S rDNA-sensor chip was investigated by monitoring the ΔC when repeatedly injecting the same concentration of the standard complementary 30-mer ss16S rDNA (10-11 M) up to 23 times in 2 days, with regeneration after each assay. The residual capacity was calculated (Eq. 1 below) and the values of the percentage residual capacity of the 30-mer 16S rDNA-sensor chip versus the number of injections.
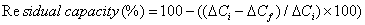 (1)
(1)
where, ΔCi is the initial capacitance change and ΔCf is the final capacitance change.
Stock solution of bacterial cells: E. coli was cultivated on LB- agar plates at 37°C for 24 hr., while L. reuteri cell mass was provided by Dr. Tarek Dishisha (Department of Biotechnology Lund, University). The cells of E. coli and L. reuteri were harvested by spinning down the cells at 3000 × g for 5 min. The cells were thereafter re-suspended in a saline solution (0.9% (w/v) NaCl) and kept at 4°C until further use. The number of cells per ml of each culture was determined microscopically using a hemocytometer [18] just before use.
DNA sample preparation from bacterial cells: It should be noted that, both E. coli and L. reuteri samples were treated in a similar manner throughout the study.
The whole sample preparation took less than 90 min and the hybridization time was 15 min. The preparation started by (1) Whole genomic DNA extraction (2) DNA fragmentation by restriction enzyme (3) DNA heat denaturation (4) Preventing re-annealing of single stranded DNA by adding ET SSB and (5) Injecting DNA sample into the capacitive DNA-sensor for analysis, immediately after adding formamide to 30% (v/v), as shown in Figure 1.

Figure 1: Schematic diagram of DNA sample preparation for capacitive DNA-sensor analysis.
DNA extraction: Genomic DNA were extracted from E. coli and L. reuteri cultures (about 108 cells ml-1) using ZR Fungal/Bacterial DNA MiniPrepTM. The concentration of the extracted genomic DNAs was estimated using a NanoDrop spectrophotometer.
DNA fragmentation: AciI (SsiI) restriction enzyme was used as described in the protocol (Fermentas) for digestion of genomic DNA. Inorder to produce suitable and consistent targeted fragments, the digestive efficiency of AciI restriction enzyme was previously studied. Whereas the 16S rDNA was initially amplified from 2 μL E. coli DNA extract with 10 μL Dream Taq polymerase (dNTPs and polymerase), the set of primers, 1 μL 63F and 1 μL 1386R, and 6 μL water; to make a PCR reaction mixture of 20 μL. PCR was perfomed for 30 cycles, starting at initial denaturation temperature, 95°C for 3 min, at the first cycle, for the consecutive 29 cycles the same denaturation temperature was kept for 30 s. The annealing and extension temperatures were 50°C for 30 s and 72°C for 2.5 min throughout the PCR run, respectively. However, for the final step, the extension temperature was kept for 7.5 min. Then, the PCR product was digested using AciI (SsiI) enzyme.
Both, whole PCR product and fragments were analysed by electrophoresis on 3% agarose gel, run at 80 V for 2 hr. The gel was stained in GelRedTM 3X staining solution and then visualized and photographed with MiniBis Pro 16 mm from DNR Bio-Imaging system.
Heat denaturation of DNA: In order to produce single stranded DNA for DNA-sensor analysis, a mixture which contained: 50 μL of 14 ng μL-1 fragmented DNA sample, 29 μL nuclease free water, and 20 μL 5X Phusion buffer was heated at 95°C for 5 min. Then, 1 μL (0.5 μg) of ET SSB was added to the mixture while heated in order to prevent re-annealing of denatured DNA. Finally, the sample was ready for analysis.
The effect of addition of ET SSB was previously compared with a rapid-cooling approach for preventing re-annealing of denatured DNA, whereas three different cooling media were studied (Figure 2).

Figure 2: Schematic diagram: Approach to study the effect of different conditions of DNA heat denaturation in the stability of ssDNA.
Two complementary single stranded 30-mer 16S rDNAs were mixed at 1:1 ratio and heated on a water bath, 60°C for 5 min and left to hybridize at room temperature for 1 h. Six aliquotes of 100 μL each; named 2, 3, 4, 5, 6 and 7 were taken from the reaction mixture. The aliquotes and a 100 μL single stranded 30-mer 16S rDNA control sample solution (named 1), were then heated in the water bath at 95°C for 5 min; while heated, total volume of two microliter ET SSB solution was added in to samples 6 and 7, one microliter each. After the heating, samples 1 and 2 were left at room temperature (23°C), samples 6 and 7 were incubated at 37°C; samples 3, 4, and 5 were immediately transferred to ice water (0.6°C), a cryostat (-12°C, ethanol as cryo-solvent), and dry ice (-78°C), respectively. All samples were left at their respectively temperatures for 1 h. After that, in sample 7 about 30 μL of formamide was added, mixed and the mixture incubated at room temperature for 5 min. All samples were then analysed with gel electrophoresis using 3% agarose gel, 80 V for 2 h 45 min.
E. coli sample analysis
Capacitive DNA-sensor: Since the initial concentration used for extraction of genomic DNA was 108 cells ml-1, it was considered that the extract contained a concentration of genomic DNA corresponding to the concentration of the starting amount the cells (108 cells ml-1). It was considered that 100% recovery of genomic DNA was achived using ZR Fungal/Bacterial DNA MiniPrepTM extraction kit under optimum conditions. Moreover, following dilutions during the DNA fragmentation and denaturation steps, the genomic DNA ended up being 10 times diluted. Prior to DNA-sensor analysis, the pre-treated genomic DNA was even further serially diluted to 1/103, 1/104, 1/105, 1/106, 1/107 and 1/108. Based on the assumption above, these dilutions were assumed to correspond to concentrations of 105, 104, 103, 102, 101 and 1 E. coli cell(s) ml-1, respectively. The assumption above was made for the sake of comparison between capacitive DNA-sensor and the plate counting techniques. Finally, an aliquot of each dilution was injected into the capacitive DNA-sensor in triplicate, and ΔC for each dilution was determined. Sensitivity of the sensor was then compared with the plate counting technique.
Plate counting: Five different concentrations of E. coli cells: 101, 102, 103, 104 and 105 cells ml-1 were prepared by serially diluting a 108 cells ml-1 stock suspension by a factor of 10. Each concentration above was treated as an individual sample. The LB agar plates in triplicates were then streaked with 10 μl of each sample and incubated at 37°C for 24 h. Samples with 103 to 105 cells ml-1 were initially diluted to obtain an acceptable range of colony forming unit per plate (CFU/ plate). Finally, the CFU ml-1 of each sample was determined [19].
Selectivity of the DNA-sensor chip
Whole genomic DNA: An aliquot of each dilution of L. reuteri (family Lactobacillaceae) genomic DNA were analysed by the capacitive DNA-sensor. The analyses were done in triplicate, and ΔC for each dilution was determined and compared with that of the E. coli (family Enterobacteriacea) genomic DNA. The experiment was repeated by injecting samples of E. coli and L. reuteri at a dilution corresponding to 105 cells ml-1, while changing the working temperature from initially 30 to 40°C, but keeping the same concentration of formamide in the mobile phase.
Amplified 16S rDNA: Genomic DNAs of E. coli and L. reuteri samples were initially extracted as explained in DNA extraction section, and then 16S rDNA from diluted DNA extract (1:10) of each sample was amplified with Dream Taq PCR master mix and the set of primers 63F and 1386R. PCR was perfomed for 30 cycles (in the same way as in DNA fragmentation section). PCR products of each sample were analysed on 1% agarose gel, run at 120 V for 55 min. A band for 16S rDNA from each sample was carefully excised, and then 16S rDNA was extracted from the gel using Omega Bio-tek’s gel extraction kit. Equal volume of 10-10 M 16S rDNA samples from L. reuteri and E. coli separately, were enzymatically fragmented and heat-denatured as described in DNA fragmentation section and Heat denaturation of DNA section, respectively. Thereafter, each sample was injected into the capacitive DNA-sensor system, which was previously modified by immobilizing a synthesized 30-mer E. coli 16S rDNA. The ΔC for each sample was determined and compared to each other.
Results and discussion
Modification of sensor electrode surface
Cyclic voltammogram of each electrode surface modification was recorded as shown in Figure 3 and compared with a cyclic voltammogram of a bare electrode surface (insert, a).

Figure 3: Cyclic voltammograms recorded in 10 mM K03[Fe(CN)6] in 0.1M KCl. The potential was swept in the range between -0.25 and 0.7 V (vs Ag/ AgCl) with a sweep rate of 0.1 V s-1: an inset (a) bare gold electrode (blue); (b) gold electrode modified with polytyramine layer (red); (c) the same as (b) after immobilization of 30-mer E. coli 16Ss rDNA (black); (d) as in (c) after treatment with 1-dodecanethiol (green).
The electrode surface was initially functionalized with amine groups via the polytyramine layer that was electro-deposited onto the electrode surface. The dramatic decrease in redox peak currents after deposition of polytyramine layer (b, red) reflects the passivation of the electrode surface, because the formed non-conducting layer hampers the electron transfer between electrode surface and redox species. Following immobilization of the capture probe onto the surface, the redox peak currents were further decreased (c, black). Finally, a pinhole-free surface was formed by depositing 10 mM 1-dodecanethiol (in ethanol) on the surface, which resulted in an even further decrease in redox peak currents (d, green).
Capacitive DNA-sensor for assay of hybridization of complementary probes
Linear dynamic range and detection limit: The fundamental principle of the capacitive transducer is based on the theory of double layer (DL) which was well described previously by Berggren et al. [20]. During the capacitance measurement, charge transfer is taking place at the DL [15]. The modified gold electrode when placed in an electrolyte solution (PB), behaves like several capacitors arranged in series (Figure 4) with; Cins capacitance of the insulating layer on the surface; Ccp capacitance of immobilized capture probe; and Cdl capacitance of the double layer..

Figure 4: Schematic drawing of different layers covering the electrode surface comprising the total capacitance (Ctot), Cins is the insulating layer capacitance; Ccp, the capture probe capacitance; and Cdl, the double layer capacitance.
Therefore, the total capacitance (Ctot) of the modified electrode is defined by Eq. (2):
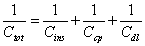 (2)
(2)
When target ssDNA hybridizes to the capture probe, the dielectric constant of the electrode surface layer will decrease [21], and the double layer will be pushed away from the electrode surface [20]. The decrease in the dielectric constant and the displacement of the double layer will result in the decrease of relative permittivity of the electrode/ solution interface, and in an increase of the distance between ions and the electrode surface layer, respectively. The former will result in the decrease of Ccp, whereas, the latter will cause the decrease of Cdl; and therefore, both will result in the decrease of the Ctot registered.
Here, the change in capacitance i.e., the difference of the Ctot before and after sample injection (ΔC) was found to be proportionally related to the logarithmic concentrations of the target ssDNA sample. The linear dynamic range and LOD of the capacitive DNA-sensor is shown in Figure 5.

Figure 5: Capacitance change against logarithmic concentration of complementary 30-mer E. coli 16S rDNA.
The linear range vs. logarithmic concentration of capacitive DNAsensor was between 1.0 × 10-12 to 1.0 × 10-7 M, with the sensitivity of 13.6 -nF cm-2 M-1 and LOD of 6.5 × 10-13 M, determined according to IUPAC recommendation [22]. The detection limit of the capacitive DNA-sensor reported here is in agreement with Sankoh et al. [23] who reported the limit of detection of 2.0 × 10-13 M and linear range of 1.0 × 10-12 to 1.0 × 10-8 M. The obtained linear range from Figure 5 above, represents a slightly wider range than what is reported previously [23].
Reproducibility: The reproducibility of the analysis involving the 30-mer 16S rDNA-sensor chip was investigated. The residual capacity of the 30-mer 16S rDNA-sensor chip versus the number of injections is depicted in Figure 6. After the first 20 injections, the hybridization capacity of the 30-mer 16S rDNA-sensor chip retained about 88 ± 5% of the original capacity.

Figure 6: Response from a 30-mer E. coli 16S rDNA modified electrode to 23 injections of a 250 μL of standard complementary 30-mer E. coli 16S rDNA (1.0 × 10-11 M) with intermittent regeneration steps.
Thus, the 30-mer 16S rDNA-sensor chip could be re-used with good reproducibility up to 20 times (relative standard deviation (RSD) of 5.8%). After 20 times of hybridization and regeneration (0.5 M NaOH), the sensor chip hybridization capacity dramatically decreased to ca 50%. The reduction of capacity may be caused by the loss of ssDNA capture probe on the sensor chip after repeated hybridization and regeneration cycles. Another explanation for the decrease of the DNA-sensor capacity could be microbial and nuclease contamination because the assay procedure was conducted under non-sterile condition.
Assay for real bacterial DNA sample
Pre-capacitive DNA-sensor processing: A diagnostic label-free capacitive DNA-sensor could be an extremely powerful and rapid tool for clinical diagnosis of microbial infections and genetic diseases, as well as for detecting microorganisms in environmental and food samples. The usefulness of the capacitive DNA-sensor technique is limited by the ability of the single-stranded DNA to rebind after dissociation which can lead to the false-negative results even if the DNA fragment of the targeted gene is abundantly present in the sample. Therefore, DNA sample processing prior to the assay is required. In this work, a commercial kit was used to extract genomic DNA samples from E. coli and L. reuteri cultures containing 108 cells ml-1 each.
The method is based on the selective adsorption of nucleic acids to a silica-gel membrane in the presence of high concentrations of chaotropic salts as it was described elsewhere [24]. The genomic DNA was extracted in order to reduce background noise due to matrix effects of the sample by excluding other cellular components such as proteins, but also to get rid of RNA. Infact, the presence of RNA similar to the complementary targeted ssDNA probes could lead to the false-positive results. The DNA extraction step yielded genomic DNA with total concentrations of about 70 ng/μl.
The genomic DNA was fragmented to produce a suitable size of targeted complementary sequence (30-mer) with respect to the capture probe on the DNA-sensor chip. DNA fragments were produced using AciI restriction enzyme, of which according to Webcutter software AciI enzyme recognizes CCGC (-3/-1)^ sites and cuts E. coli 16S rDNA at 17 different positions, including sites 374 and 421, between which the targeted complementary sequence is found. To demonstrate this concept, the PCR product of 16S rDNA was digested with AciI restriction enzyme and the fragments were run in gel electrophoresis. The results are shown in Figure 7; the product includes the targeted complementary fragment on lane D of about 44 bp in size.

Figure 7: E. coli 16S rDNA PCR products digested with AciI restriction enzyme separated on a 3 % agarose gel, 80V, 2h: L1: Ultra low range DNA ladder; UD: Un-digested 16S rDNA; D: Digested 16S rDNA; and L 2: DNA ladder (1kb).
When a double-stranded DNA is heated close to the boiling point of water, the hydrogen bonds that bind the two strands together in a double helix structure break and the DNA is separated into two complementary strands. However, if the denatured DNA sample is allowed to cool to between 50 and 60°C, the single strands will reassociate with their complementary partners and reform a double helix. Therefore, one of the challenges in preparation for DNA-sensor analysis is to prevent the separated single stranded DNA from reannealing prior to analysis. Figure 8 represents a photograph of the electrophoretic gel, showing the results of two approaches investigated in this study by using the standard solutions of two complementary 30- mer 16S rDNAs: cooling approach; ice water (0.6°C) (lane #3), ethanol in cryostat (-12°C) (lane #4) and dry ice (-78°C) (lane #5); addition of ET SSB; (lane #6). Lane #7 is the same as lane #6 but followed by addition of formamide. All samples on the gel were run against single stranded DNA (lane #1) and double stranded DNA (lane #2) as a positive and negative control, respectively, as well as DNA ladder (lane L).

Figure 8: Electrophoresis gel profile (3% agarose, 80 V, 2 h 45 min), presenting the effect of different conditions of denaturation in the stability of ssDNA. L: DNA ladder. 1: Single stranded 30-mer 16S rDNA. 2: Double stranded 16S rDNA sample (30 bp). For lane 3 to 7: Heat denaturation (95°C) of the 16S rDNA (30 bp) was the first step of the treatment for all samples. The second step was different for each lane and only this variation is given on the legend; 3: Chilling in the ice water (0.6°C). 4: Placing in the ethanol cryostat (-12°C). 5: Chilling in the dry ice (-78°C). 6: Presence of ET SSB during the first step. 7: Like lane 6 but additional ET SSB denaturation with formamide 30%.
The principle for immediate rapid cooling of denatured DNA is to cause a rapid aggregation to the individual generated single stranded DNAs and hence, preventing them from re-annealing. The application of rapid cooling on ice water to prevent re-annealing of single stranded DNA sample was previously reported [5]. In contrast, in the present study heat denaturation (95°C, for 5 min) followed by rapid cooling did not result in detectable ssDNA on the 3% agarose gel; as depicted in Figure 8, all the bands obtained from the rapid cooling approach; water ice (lane # 3), cryostat (lane #4), and dry ice 5 (lane # 5), migrated to the same position as that for double stranded DNA sample (lane #2), and it is found between 25 and 35 bp bands on the DNA ladder (L), which is equivalent to 30 bp. Hence, the rapid cooling approach could not produce a meaningful amount of ssDNA for analysis. Also, Thompson has previously reported that, chilling solution of denatured DNA at usual concentrations of 8 - 12 mg/ml did not result in aggregate formation [25]. He further demonstrated that, DNA aggregation positively is favoured by a high concentration and substantial length of a DNA. Therefore, a possible explanation for the obtained result could be the low concentration (14 ng μL-1) and short DNA sequence (30- mer) used in this study.
The band for heat-denatured DNA sample treated with ET SSB (lane #6) has migrated to the position of the 30-mer single stranded DNA (lane #1), which is equivalent to the band of about 25 bp on the DNA ladder (L). This study has verified that the heat-denatured DNA sample can be prevented from re-annealing by the addition of ET SSB. The interactions between single stranded DNA and the binding proteins (SSBs)-DNA was previously studied using surface plasmon resonance imaging [26], whilst the mechanisms of SSBs-DNA interaction was described well by Kunzelmann et al. [27]. The SSB prevents complementary ssDNA from re-annealing by binding specifically with high affinity to single-stranded DNAs [28]. The combination of electrostatic, hydrogen-bonding and stacking interactions from SSB to ssDNA forms the basis for ssDNA binding and specificity. However, the hydrophobic nature of stacking interactions provides the SSB with an additional strategy for ssDNA binding that does not require specialization of the nucleic acid-binding surface into specific DNA binding pockets or hydrogen-bonding patterns that would favour one sequence over another [29].
Then, one needs to remove ET SSB from ssDNA sample prior to injection into the DNA-sensor to allow the hybridization with the ssDNA-capture-probe on the sensor-chip. Therefore, the sample was mixed with formamide prior to injection such that the mobile phase contained a final concentration of 30% formamide. In the same figure (Figure 8), lane 7 shows that, the band for the denatured DNA sample that was treated with ET SSB to prevent re-annealing, but later on mixed with formamide to final concentration of 30% and left to reassociate, has migrated to the same position of that for 30-mer double stranded DNA (lane #2), suggesting that addition of formamide to final concentration of 30% was enough to denature and remove ET SSB from the ssDNAs without affecting a hybridization with the complementary probe.
Sadhu and Dutta [30] have demonstrated that, the addition of formamide to a mixture of complementary ssDNA solution to final concentration of 30%, did not affect the rate of hybridization [30]. Also, Knubovets et al. [31] have studied the behaviour of a protein in organic solvents and reported that, the protein (e.g. ET SSB) unfolds in solvents like formamide which competes for backbone hydrogen bonds. However, the addition of formamide in the sample should be done immediately before applying the sample on the DNA-sensor chip, the addition of formamide will cause the re-annealing if the sample is left for long time before applying on the DNA-sensor chip such that one loses ability to see the binding to the probe on the sensor surface.
Analysis of E. coli using capacitive DNA-sensor versus plate counting: Table 2 shows the results obtained by the capacitive DNAsensor and plate counting techniques.
| Oligonucleotide | Sequence (5’? 3’) | Size |
|---|---|---|
| Capture probe | GAGTAAAGTTAATACCTTTGCTCATTGACG | 30-mer |
| Target Probe | CGTCAATGAGCAAAGGTATTAACTTTACTC | 30-mer |
| Forward primer (63F) | CAGGCCTAACACATGCAAGTC | 21-mer |
| Reverse primer (1389R) | ACGGGCGGTGTGTACAAG | 18-mer |
| E.coli 16S r-DNA (amplified sequence) | - | 1447 bp |
| Target fragment of E.coli 16S rDNA (Acil, restriction enzyme product) | GGTAACGTCAATGAGCAAAGGTA TTAACTTTA CTCCCTTCCTCC | 44-bp |
Table 1: Different oligonucleotide sequences and their sizes used in this study.
| Apllied no. of cells mI1 | Plate counting (CFU mI1) | DNA sensor (-nF cm-2) |
|---|---|---|
| 105 | (9.5 ± 0.5)×104 | 84 ± 9 |
| 104 | (7.8 ± 0.3)×103 | 57 ± 5 |
| 103 | (8.8 ± 0.2)×102 | 40 ± 4 |
| 102 | (9.2 ± 0.2)×101 | 27 ± 4 |
| 101 | ND | 21 ± 3 |
| 100 | ND | 10 ± 1* |
*Below Limit of Detection. ND = Not Detected
Table 2: Comparison: Detection of E. coli cells at concentration range 1 to 105 cells ml-1 using plate counting method and DNA based technique, capacitive DNAsensor.
The capacitive DNA-sensor proved more sensitive than plate counting, with a detection limit of 10 E. coli cells ml-1. This is a value ten times lower than that detected by the plate counting technique (100 cells ml-1). In addition, the capacitive DNA-sensor can detect both viable and dead cells from the sample; which in many aspects could be regarded as advantageous. In fact, as other pathogens such as Staphylococcus aureus, Bacillus cereus and Clostridium botulinum are capable of producing toxins in the food sample [32] and thus, actively living bacterial cells do not need to be present for food illness to develop. However, even if the bacterial cell walls already have been destructed and the cells are dead, the DNA is stable and available in the sample. Hence, pathogens will be positively detected. In addition to that, capacitive DNA-sensor provides a detection solution for the problem of the bacteria in a viable but non-culturable (VBNC) state; unlike for the plate the counting method, which would only propagate viable and cultivable cells.
This capacitive DNA-sensor is even more sensitive than other biosensors reported in the literatures; LiJiang et al. [33] and Luo et al. [34], reported detection of approximately 2000 Cfu ml-1 of E. coli O157:H7 and 40 Cfu ml-1 of Enterobacteriaceae bacteria using a gold nanoparticle-enhanced piezoelectric biosensor and exonuclease IIIenhanced- electrochemical DNA biosensor, respectively. Muhammad- Tahir and Alocilja (2003) [35] reported on an electrochemical sandwich immunoassay that could detect about 78 Cfu ml-1 of E. coli O157:H7.
Selectivity: To investigate a cross reactivity of DNA-sensor chip at a family level; L. reuteri DSM 20016 (Lactobacillaceae) was used as a candidate, and both un-amplified genomic DNA and amplified 16S rDNA were tested. According to Blast analysis on NCBI (http://blast. ncbi.nlm.nih.gov/Blast.cgi) [36], L. reuteri DSM 20016 has 45 sites on its genomic DNA that match with the capture probe. Five of these matching sites were found to have thirteen matching bases with the capture probe, that make upto 43% of the total number of bases on the capture probes. Nevertheless, among the 45 matching sites, three are from its 16S rDNA sequence and were found to have only 7 matching bases with the capture probe, about 23% of the total bases on the capture probe.
Figure 9a presents the results of the capacitance change from the hybridization of equal concentration range of un-amplified single stranded genomic DNAs of E. coli and L. reuteri. The maximum capacitance change obtained from the injection of whole genomic DNA of L. reuteri corresponded to 105 cells ml-1 is 32 ± 4 -nF cm-2, which is almost three times lower than the response obtained from the injection of non-amplified whole genomic DNA of E. coli at equal concentration (84 ± 9 -nFcm-2).

Figure 9a: Selectivity study: capacitive responses from DNA-sensor chip with immobilized 30-mer E. coli 16S rDNA, following injection of unamplified whole genomic DNA samples at concentration range corresponding to 101 to 105 cell ml-1 of E. coli and L. reuteri, at working (hybridization) temperature of 30°C, and 30% formamide in the mobile phase.
The response of L. reuteri (7-nF cm-2) was 3 times lower than the response of E. coli genomic DNA (21 ± 3 -nF cm-2) corresponding to concentration of 101 cells ml-1, which approaches the detection limit of the technique, indicating that the developed DNA chip has high selectivity to the E. coli DNA at the given working conditions: 30°C working temperature and 30% formamide added in the mobile phase. Several authors have reported the non-specific hybridization between capture probe and non-perfectly matched DNA probe [20,23,37-39].
However, the selectivity of the DNA-sensor chip was further studied at elevated working temperature of 40°C, while keeping the same formamide concentration (30%) in the mobile phase, by injecting the un-amplified genomic DNA of L. reuteri and E. coli at a DNA concentration that is corresponding to 105 cell ml-1. Similarly, the experiment was repeated at 30°C, while keeping other conditions the same. The results obtained from both working temperatures are compared as shown in Figure 9b.

Figure 9b: Selectivity study: capacitive responses from DNA-sensor chip with immobilized 30-mer E. coli 16S rDNA, following injection of unamplified whole genomic DNA samples at concentration range corresponding to 101 to 105 cell ml-1 of E. coli and L. reuteri, at working (hybridization) temperature of 30°C, and 30% formamide in the mobile phase.
The capacitance change obtained from injecting L. reuteri was efficiently reduced from, 32-nF cm-2 (at 30°C), to 0.14 -nF cm-2 at the elevated working temperature of 40°C, without significantly altering the specific interaction of the applied E. coli BL21(DE3) genomic DNA. The responses due to specific hybridizations are 84 and 78-nF cm-2 for working temperatures of 30 and 40°C, respectively. Besides, the observed non-specific signal (0.14 -nF cm-2) at working temperature of 40°C, was below the lowest detection limit of the system. In particular, the hybridization was performed at very high stringency conditions; in other words, the presence of 30% formamide in the mobile phase, lowers the melting temperature of the complementary probes from 57.5°C to 35.9°C; since for every 1% of formamide reduces the melting temperature by 0.72°C [30,40,41], consequently, raising the working temperature to 40°C, makes the hybridization temperature to be slightly higher than the melting temperature. These results suggest that one could discriminately detect E. coli by injecting its whole genomic DNA in the presence of whole genomic DNA of L. reuteri at 40°C hybridization temperature, and 30% formamide in the mobile phase, without involving any DNA amplification step.
To demonstrate that the non-selectivity observed at 30°C and 30% formamide was due to other genes on the whole L. reuteri DSM 20016 genomic DNA (with matching bases are more than 7), rather than its 16S rDNA sequence, the 16S rDNAs of E. coli and L. reuteri were each amplified and purified. Thereafter, the DNA material was injected in the capacitive DNA-sensor, at working temperature 30°C containing 30% formamide in the mobile phase. The results are shown in Figure 9c.

Figure 9c: Selectivity study after DNA amplification: capacitive responses from DNA-sensor chip with immobilized 30-mer E. coli 16S rDNA; following injection of 10-10 M amplified 16S rDNA samples from E. coli and L. reuteri cells, at working (hybridization) temperature of 30°C, and 30% formamide in the mobile phase.
The capacitive DNA-sensor showed no significant responses towards L. reuteri 16S rDNA samples; on the other hand for E. coli 16S rDNA sample, a clear and significant signal was observed.
These results indicate that, the non-specific signals that were observed when the whole genomic DNA of L. reuteri was applied, were due to hybridization of the capture probe with other genes on the L. reuteri genomic DNA, probably those five genes with 13 matching bases to capture probes.
When whole genomic DNA is used for selective identification of a target bacteria based on its 16S rDNA (for example the hyper variable region), a capture probe should be design in such that, it does not match with 16S rDNA or any part of the genomic DNA of other bacteria. This may be a complex task, especially when a designed capture probe is as short as 15-30 mer. Particularly, since the shorter the sequence of the capture probe the higher probability of non-selectivity can be expected [20]. Otherwise, working close to the melting temperature of the capture probe could be the best alternative approach. Most of the sensor flow cell materials however do not withstand the temperature above 45°C [20]. Hence, the addition of formamide in the mobile phase has been demonstrated in this study, eliminating the need of equipment that tolerates higher temperature. Formamide reduces the thermal stability of DNA probes and hence, allows operation at suitable temperature for the sensor set ups with higher selectivity.
Conclusion
We have reported a flow-based ultrasensitive PCR-free capacitive DNA-sensor for detecting the complementary sequence from unamplified Enterobacteriaceae genomic DNA. The reported sensor was relatively fast, 15 min hybridization time; and tenfold more sensitive than plate counting, and even more sensitive than other biosensors reported in the literatures; [33-35]. Also, for the first time we have demonstrated the alternative approach to bacterial DNA sample preparation for flow-based DNA analysis. The approach involved using an extreme thermostable ssDNA binding protein (ET SSB), to prevent re-annealing of a heat-denatured target ssDNA prior to analysis; and during analysis formamide integrated in the running buffer to denature ET SSB. In addition, the DNA-sensor chip could be reused with reproducibility up to 20 times with a RSD of <6%. The DNAsensor chip has shown selectivity for Enterobacteriaceae DNA over Lactobacillaceae DNA at elevated working temperature and presence of upto 30% formamide in the running buffer. Thus, it is practical to develop a PCR-free, sensitive and selective flow-based capacitive DNAsensor for bacteria detection via specific DNA analysis.
The reported capacitive DNA-sensor has potential for several applications in the health care sector, in food/water quality control and in research institute laboratories. In addition, the reported DNA-sensor lays the groundwork for incorporating the method into an integrated system for in-field bacteria detection. However, in order to be able to use the DNA-sensor at physiological level, one needs to have a full understanding of the electrochemical and topographical properties of the surface of the DNA-sensor chip. Therefore, future work will be needed to give the necessary insight of electrode surface roughness properties, and packing arrangement of the insulating matrices and capture probes.
Acknowledgements
Financial support from World Bank supported project titled “Capacity Building in Science, Technology and Higher education” at University of Dar es Salaam, Tanzania, is gratefully acknowledged. Also, technical and logistical support from Higher education and Swedish Research Council is highly appreciated.
References
- Janda JM, Abbott SL (2002) Bacterial identification for publication: when is enough enough? J Clin Microbiol 40: 1887-1891.
- Taege AJ (2002) The new American diet and the changing face of foodborne illness. Cleve Clin J Med 69: 419-424.
- Madigan MT, Martinko JM, Stahl DA, Clark DP (2012) Brock Biology of Microorganisms. 13th edition, Pearson, San Francisco.
- Weenk GH (1992) Microbiological assessment of culture media: comparison and statistical evaluation of methods. Int J Food Microbiol 17: 159-181.
- Arora K, Prabhakar N, Chand S, Malhotra BD (2007) Escherichia coli genosensor based on polyaniline. Anal Chem 79: 6152-6158.
- Jiang D, Liu F, Liu C, Liu L, Pu X (2013) An Electrochemical Sensor Based on Allosteric Molecular Beacons for DNA Detection of Escherichia coli. O157:H7. Int J Electrochem Sci8: 9390-9398.
- Rijal K, Mutharasan R (2013) A method for DNA-based detection of E. coli O157:H7 in a proteinous background using piezoelectric-excited cantilever sensors. Analyst 138: 2943-2950.
- Patel MK, Solanki PR, Kumar A, Khare S, Gupta S, et al. (2010) Electrochemical DNA sensor for Neisseria meningitidis detection. Biosens Bioelectron 25: 2586-2591.
- Pang S, Gao Y, Li Y, Liu S, Su X (2013) A novel sensing strategy for the detection of Staphylococcus aureus DNA by using a graphene oxide-based fluorescent probe. Analyst 138: 2749-2754.
- Hsu SH, Lin YY, Lu SH, Tsai IF, Lu YT, et al. (2013) Mycobacterium tuberculosis DNA detection using surface plasmon resonance modulated by telecommunication wavelength. Sensors (Basel) 14: 458-467.
- Naravaneni R, Jamil K (2005) Rapid detection of food-borne pathogens by using molecular techniques. J Med Microbiol 54: 51-54.
- Ye Y, Ju H (2003) DNA Electrochemical behaviours, Recognition and Sensing by Combining with PCR Technique. Sens 3: 128-145.
- Rådström P, Knutsson R, Wolffs P, Lövenklev M, Löfström C (2004) Pre-PCR processing: strategies to generate PCR-compatible samples. Mol Biotechnol 26: 133-146.
- Jarzab A, Gorska-Fraczek S, Rybka J, Witkowska D (2011) Enterobacteriaceae infection - diagnosis, antibiotic resistance and prevention. Postepy Hig Med Dosw 65: 55-72.
- Mahadhy A, Ståhl-Wernesson E, Mattiasson B, Hedström M (2014) Use of capacitive affinity biosensor for sensitive and selective detection and quantification of DNA- A model study. Biotechnol Rep 3: 42-48.
- Teeparuksapun K, Hedström M, Wong EY, Tang S, Hewlett IK, et al. (2010) Ultrasensitive detection of HIV-1 p24 antigen using nanofunctionalized surfaces in a capacitive immunosensor. Anal Chem 82: 8406-8411.
- Tichoniuk M, Ligaj M, Filipiak M (2008) Application of DNA Hybridization Biosensor as a Screening Method for the Detection of Genetically Modified Food Components. Sens 8: 2118-2135.
- Strober W (2001) Monitoring cell growth. Curr Protoc Immunol.
- Sutton S (2011) Accuracy of Plate Counts. J Valid Technol 17: 42-46.
- Berggren C, Bjarnason B, Johansson G (2001) Review: Capacitive Biosensors. Electroanal 13: 173-180.
- Rivera-Gandia J, Cabrera CR (2007) Self-assembled monolayers of 6-mercapto-1-hexanol and mercapto-n-hexyl-poly(dT)18-fluorescein on polycrystalline gold surfaces: An electrochemical impedance spectroscopy study. Electroanal Chem 605: 145-150.
- Buck RP, Lindner E (1994) Recommendations for Nomenclature of Ion-selective Electrodes. Pure Appl Chem 66: 2527-2536.
- Sankoh S, Samanman S, Thipmanee O, Numnuam A, Limbut W, et al. (2013) A comparative study of a label-free DNA capacitive sensor using a pyrrolidinyl peptide nucleic acid probe immobilized through polyphenylenediamine and polytyramine non-conducting polymers. Sens Actuators B 177: 543-554.
- Melzak KA, Sherwood CS, Turner RFB, Haynes CA (1996) Driving Forces for DNA Adsorption to Silica in Perchlorate Solutions. J Colloid Interface Sci 181: 635-644.
- Thompson WF (1976) Aggregate formation from short fragments of plant DNA. Plant Physiol 57: 617-622.
- Brockman JM, Frutos AG, Corn RM (1999) A Multistep Chemical Modification Procedure to Create DNA Arrays on Gold Surfaces for the Study of Protein-DNA Interactions with Surface Plasmon Resonance Imaging. J Am Chem Soc 121: 8044-8051.
- Kunzelmann S, Morris C, Chavda AP, Eccleston JF, Webb MR (2010) Mechanism of interaction between single-stranded DNA binding protein and DNA. Biochemistry 49: 843-852.
- Jain S, Zweig M, Peeters E, Siewering K, Hackett KT, et al. (2012) Characterization of the single stranded DNA binding protein SsbB encoded in the Gonoccocal Genetic Island. PLoS One 7: e35285.
- Shamoo Y (2001) Single-stranded DNA-binding Proteins. eLS.
- Sadhu C, Dutta S, Gopinathan K (1984) Influence of formamide on the thermal stability of DNA. J Biosci 6: 817-821
- Knubovets T, Osterhout JJ, Klibanov AM (1999) Structure of lysozyme dissolved in neat organic solvents as assessed by NMR and CD spectroscopies. Biotechnol Bioeng 63: 242-248.
- Adams MR, Moss MO (2008) Food Microbiology. 3rd Edition, Royal Society of Chemistry, Cambridge.
- LiJiang W, QingShan W, ChunSheng W, Zhao-Ying H, Jian J, et al. (2008) The Escherichia coli O157:H7 DNA detection on a gold nanoparticle-enhanced piezoelectric biosensor. Chin Sci Bull 53: 1175-1184.
- Luo C, Tang H, Cheng W, Yan L, Zhang D, et al. (2013) A sensitive electrochemical DNA biosensor for specific detection of Enterobacteriaceae bacteria by Exonuclease III-assisted signal amplification. Biosens Bioelectron 48: 132-137.
- Muhammad-Tahir Z, Alocilja EC (2003) Fabrication of a Disposable Biosensor for Escherichia coli O157:H7 Detection. Sens J IEEE 3: 345-351.
- NCBI (2013) Basic Local Alignment Search Tool.
- Carrara S, Gürkaynak FK, Guiducci C, Stagni C, Benini L, et al. (2007) Interface Layering Phenomena in Capacitance Detection of DNA with Biochips. Sens Transducers J 76: 969-977.
- Tombelli S, Minunni M, Mascini M (2005) Piezoelectric biosensors: Strategies for coupling nucleic acids to piezoelectric devices. Methods 37: 48-56.
- Yu CJ, Wan Y, Yowanto H, Li J, Tao C, et al. (2001) Electronic detection of single-base mismatches in DNA with ferrocene-modified probes. J Am Chem Soc 123: 11155-11161.
- Yilmaz LS, Loy A, Wright ES, Wagner M, Noguera DR (2012) Modeling formamide denaturation of probe-target hybrids for improved microarray probe design in microbial diagnostics. PLoS One 7: e43862.
- McConaughy BL, Laird CD, McCarthy BJ (1969) Nucleic acid reassociation in formamide. Biochemistry 8: 3289-3295.
Relevant Topics
Recommended Journals
Article Tools
Article Usage
- Total views: 15650
- [From(publication date):
November-2014 - Aug 29, 2025] - Breakdown by view type
- HTML page views : 10950
- PDF downloads : 4700