 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Our Group organises 3000+ Global Conferenceseries Events every year across USA, Europe & Asia with support from 1000 more scientific Societies and Publishes 700+ Open Access Journals which contains over 50000 eminent personalities, reputed scientists as editorial board members.
Open Access Journals gaining more Readers and Citations
700 Journals and 15,000,000 Readers Each Journal is getting 25,000+ Readers
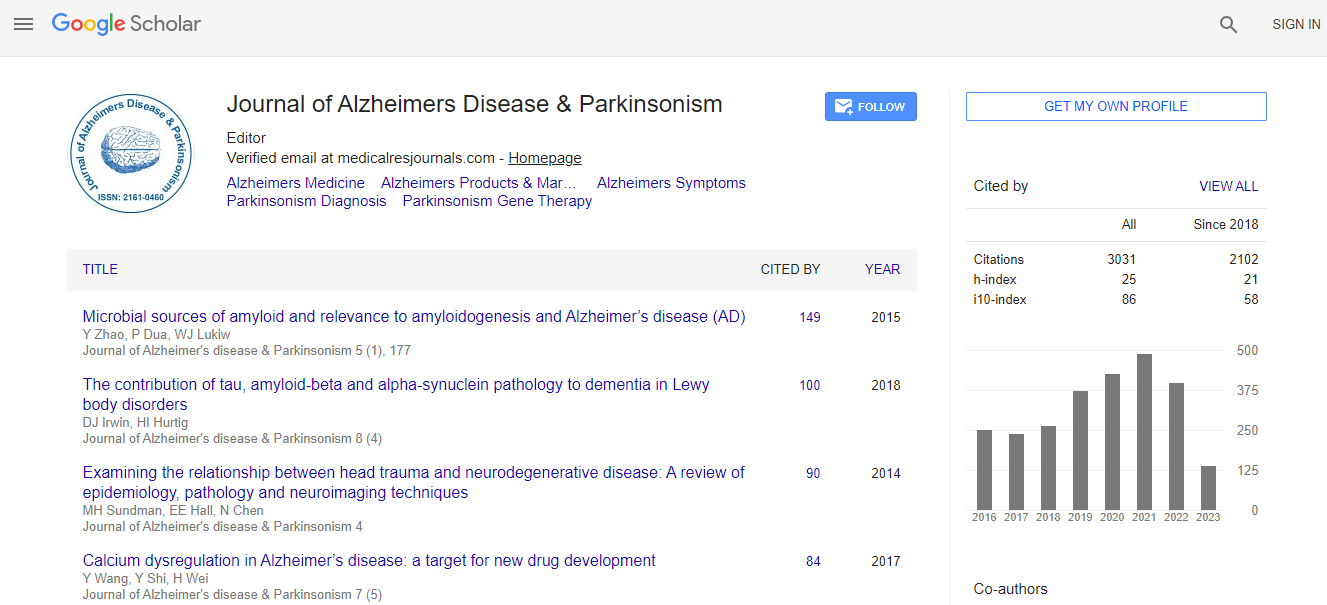
Google Scholar citation report
Citations : 4334
Journal of Alzheimers Disease & Parkinsonism received 4334 citations as per Google Scholar report
Journal of Alzheimers Disease & Parkinsonism peer review process verified at publons

Indexed In
- Index Copernicus
- Google Scholar
- Sherpa Romeo
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- JournalTOCs
- China National Knowledge Infrastructure (CNKI)
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- ICMJE
Useful Links
Recommended Journals
Related Subjects
Share This Page
Deficiency of GM1 ganglioside as major risk factor for idiopathic ParkinsonГўВ?В?s disease: Restoration of GM1-GDNF interaction in GM1 replacement therapy for PD mouse
International Conference on Parkinsons Disease & Movement Disorders
Robert W Ledeen
Keynote: J Alzheimers Dis Parkinsonism
Abstract
Several investigators have employed GM1 ganglioside to treat animal models of Parkinson’s disease (PD), and a recent clinical
trial showed PD patients treated with GM1 had lower UPDRS motor scores than at baseline after 120 weeks. Our studies of
mice with disrupted B4galnt1 gene demonstrated PD symptoms due to deficiency of ganglio-series gangliosides. Both knockout
and heterozygous (HT) mice showed depletion of striatal dopamine (DA), loss of TH+ nigral neurons, and aggregation of
alpha-synuclein. These manifestations of parkinsonism were largely alleviated by LIGA20, a membrane permeable analog of
GM1, and also by GDNF (via AAV2), essential for survival of catecholaminergic neurons. Immunohistochemical analysis of
substantia nigra sections revealed significant GM1 deficiency in TH+ nigral neurons of PD patients. Study of the occipital
cortex and colon tissue also revealed significantly lower GM1 in PD, suggesting systemic GM1 deficiency as risk factor in
idiopathic PD (Hadaczek et al. 2014). That study demonstrated GM1 association with GFRα1 and Ret, components of the
GDNF receptor, and GM1 essentiality for cohesion of those receptor proteins. Nigral neurons of PD brain showed deficient
GDNF signaling. We propose the above HT mouse with partial GM1 deficiency (similar to PD) as an especially useful PD model
in reflecting its actual pathophysiology. This is supported by detection of 3 non-movement disorders in HT mice characteristic
of PD: (a) gastrointestinal pathology (constipation), (b) cardiac sympathetic denervation, and (c) cognitive impairment. These
too were alleviated by LIGA20, further suggesting the utility of GM1 and especially its membrane permeable analogs for GM1
replacement therapy. [Supported by NIH grant RO1 NS33912].
Biography
Robert Ledeen completed his PhD at the age of 25 years at Oregon State University and postdoctoral studies at the University of Chicago and Albert Einstein
College of Medicine. He is the director of Division of Neurochemistry and a Neuroscience professor at Rutgers, New Jersey Medical School. He has published more
than 179 papers, book chapters and review articles in reputed journals and has served as an editorial board member on a number of journals.