Review Article Open Access
Tuberculosis Vaccines: Review of Current Development Trends and Future Challenges
Andrew J. Graves and David A. Hokey*
Aeras, Rockville, MD, USA
- *Corresponding Author:
- David A. Hokey
Aeras, 1405 Research Boulevard, Ste 110
Rockville, MD 20850, USA
Tel: 240-599-3077
E-mail: dhokey@aeras.org
Received Date: August 14, 2011; Accepted Date: November 18, 2011; Published Date: November 22, 2011
Citation: Graves AJ, Hokey DA (2011) Tuberculosis Vaccines: Review of Current Development Trends and Future Challenges. J Bioterr Biodef S1:009. doi:10.4172/2157-2526.S1-009
Copyright: © 2011 Graves AJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Visit for more related articles at Journal of Bioterrorism & Biodefense
Abstract
Despite plaguing humans for thousands of years, tuberculosis remains a widespread and lethal public health
problem throughout the world today. The recent rise of multi-drug-resistant tuberculosis (MDR-TB) perpetuates the public health threat while presenting a potential bioterrorism agent. The BCG vaccine is the only available prevention against TB, yet it elicits inconsistent protection when given to infants, fails to provide consistent protection in adults against pulmonary disease, and is unsafe for use in immunocompromised patients. A new crop of TB vaccine candidates has entered into clinical trials, with a second generation following shortly. These new TB vaccines are hoped to provide a safe, efficacious replacement, or addition to, the nearly century-old BCG and provide protection against TB disease beyond childhood. This review details the status of the most promising TB vaccine candidates in development, as one of these candidates may play a key role in defending against an ominous health threat.
Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, has been a threat to humanity since antiquity [1]. Despite its widespread presence throughout the world, TB is generally treatable with antibiotics [2]. While these antibiotic treatments have reduced the incidence, morbidity, and mortality of TB in developed countries such as the United States, it has also caused research for preventative alternatives to be neglected for many years. In turn, this neglect has left a large population, including the majority of U.S. citizens, vulnerable to infection. Additionally, over-reliance on antibiotics has led to development of multi-drug-resistant TB (MDR-TB) [3] and extensively drug-resistant TB (XDR-TB) [4]. Not only do these new strains represent an ominous public health threat to all nations, but MDR-TB, due to its lethality, ease to obtain, and ease of transmission was previously identified by the Centers for Disease Control as a Category C bioterrorism agent [5].
The rise of drug-resistant strains of tuberculosis has emphasized the need for better treatments and vaccines for the disease. Current drug therapies are expensive and require months of treatment. For example, the CDC recommends a total of 238 doses from a combination of three different antibiotics administered over a minimum of six months [6]. This lengthy regimen makes it difficult to ensure patient adherence, and not adhering to the full course of treatment may give further rise to MDR- and XDR-TB strains. While the expense of the antibiotic treatment is a challenge for individuals, the challenge increases exponentially when considered as part of a response to a bioterrorism event. This scenario becomes more severe should a MDR or XDR strain of TB be utilized for bioterrorism. In such cases, antibiotic therapy would be of only limited use, if at all useful. While only a small percentage of individuals exposed to TB develop active disease, this number could greatly increase during a purposeful exposure where bacterial numbers could be much higher. In addition, the fear associated with such an attack, particularly in the absence of an effective vaccine for pulmonary disease, could have a significant impact on society. While new antibiotic therapies are in development, the likelihood of antibiotics leading to a significantly improved cure in the near future seems minimal. Though TB and its antibioticresistant strains lack the sudden and dramatic effects of other potential bioterrorism agents, its ability to damage the public health system and strain national and international resources remains profound.
The best hope to counter the tuberculosis threat is development of a new vaccine. There are a number of vaccination strategies that could be employed to meet this goal. The first strategy would be to create a boosting vaccine to work in tandem with the existing BCG vaccine and supplement its immune response to prevent infection (more on BCG below). Another strategy is to develop an entirely new priming vaccine to replace BCG and improve disease prevention. Finally, vaccine candidates can also be designed as an immunotherapy, augmenting the antibiotic regimen for infected individuals and reducing recovery time. With each of these strategies having merit, there is a renewed focus for a number of research organizations to develop improved TB vaccines using modern vaccine platforms and technologies. Although this initiative is global in nature, the goal is common: enhance or replace the aging BCG vaccine which is clearly inadequate for controlling the global TB public health problem.
The BCG Vaccine
Bacille Calmette-Guérin (BCG), an attenuated strain of Mycobacterium bovis, was developed by Albert Calmette and Camille Guérin at the Pasteur Institute in the early 1900’s, with the first human vaccination in 1921 [7]. Since its introduction, BCG has proven to be relatively safe in immune-competent patients and is the most widely used vaccine in the world [8].
Despite its long history, BCG has been shown to be quite variable in its efficacy [9-11]. Researchers generally agree that, especially in infants, BCG has shown to be protective against miliary TB and TB meningitis. However, BCG recipients may still be vulnerable to pulmonary TB infection, which is the most prevalent type and causes human-tohuman transmission [12]. Another disadvantage to BCG is that infants infected with HIV, are prone to a disseminated BCG infection and thus are not recommended for vaccination [13,14], leaving them completely unprotected from the disease. BCG protection also wanes over time [15,16]. Furthermore, BCG renders no protection if administered following exposure to environmental mycobacteria [17] or Mtb [18], rendering it useless as a booster vaccine or as a priming vaccine in postexposure adults.
Mechanistically, BCG is also significantly different in its infection life cycle. Following infection into a host macrophage, mycobacteria are able to prevent phagosome-lysosome fusion [19]. On one hand, Mycobacterium tuberculosis escapes into the cytosol where it replicates [20]. Conversely, BCG is unable to escape into the cytosol. This may represent a deficiency in the BCG vaccine, as cytosolic escape may be significant for the induction of class I presentation.
In light of the development of MDR-TB and, later, XDR-TB, it became clear that improvements beyond BCG were required for TB prevention. Researchers have set out with two goals in mind: improve the efficacy of a TB vaccine against pulmonary TB, and provide a safe vaccine to immunocompromised patients.
Correlates of Protection – An Unanswered Question
As with HIV, progress within TB vaccine development is hinderedby the lack of a correlate of protection or a correlate of immunity and the path forward is not entirely clear. For example, many licensed vaccines are designed to elicit antibody responses that can either neutralize viruses or target bacteria to phagocytic cells to facilitate their destruction. Development of such vaccines then becomes a matter of inducing said antibodies. However, no such benchmark exists in the field of TB. It remains unknown whether an antibody response to Mtb would facilitate infection or help to trigger bactericidal mechanisms that would lead toward clearance.
In addition, cellular immune responses, as far as they have been currently characterized, have proven to be necessary if not sufficient. Despite these issues, there are clues that may shed light on what immune responses may be useful in fighting this disease. One of the biggest clues has come from HIV infection. HIV+ patients are many times more likely to contract TB compared to their HIV- counterparts. While there is not a definitive correlation, as there can be numerous immunological defects in HIV+ patients, there is a strong suggestion that CD4+ T cells may play a critical role in either protection against or control of TB infection. In addition, depletion studies in mice indicate that CD4+ T cells play a pivotal role in TB immunity [21].
CD8+ T cells are also implicated in control of TB. CD8+ T cell depletion in mice [22] and nonhuman primates [23] suggests CD8+ T cells are critical for host defense against TB. Despite this implication, the exact role that CD8+ T cells play in human tuberculosis infection is not clear, and further studies are required.
Th1 responses have also been implicated in the control of Mtb. The most critical of these Th1 responses appear to be Interferon-gamma (IFN-γ) [24,25] and Tumor Necrosis Factor-alpha (TNF) [26-28]. In particular, several studies have linked T cells capable of secreting multiple Th1 cytokines (generally, IFN-γ, TNF, and IL-2), known as polyfunctional T cells, with Mtb clearance [29,30]. Once again, though these types of responses are suggestive of protection, a true correlate has yet to be defined. Unfortunately, it appears now that only long-term human clinical trials will be able to determine this important endpoint
Together, these findings have been vital factors in driving TB vaccine development for the past decade, coupled with the desire for a vaccine platform that is safe and effective for all patients. The result of this ongoing vaccine development initiative is promising, with several first-generation vaccines reaching Phase IIb human clinical trials, and a crop of second-generation vaccines undergoing preclinical assessments and preparing to enter Phase I trials (Table 1). This review will focus on the some of the most promising TB vaccine candidates and the antigens they target (Table 2) with a short summary and development history of each
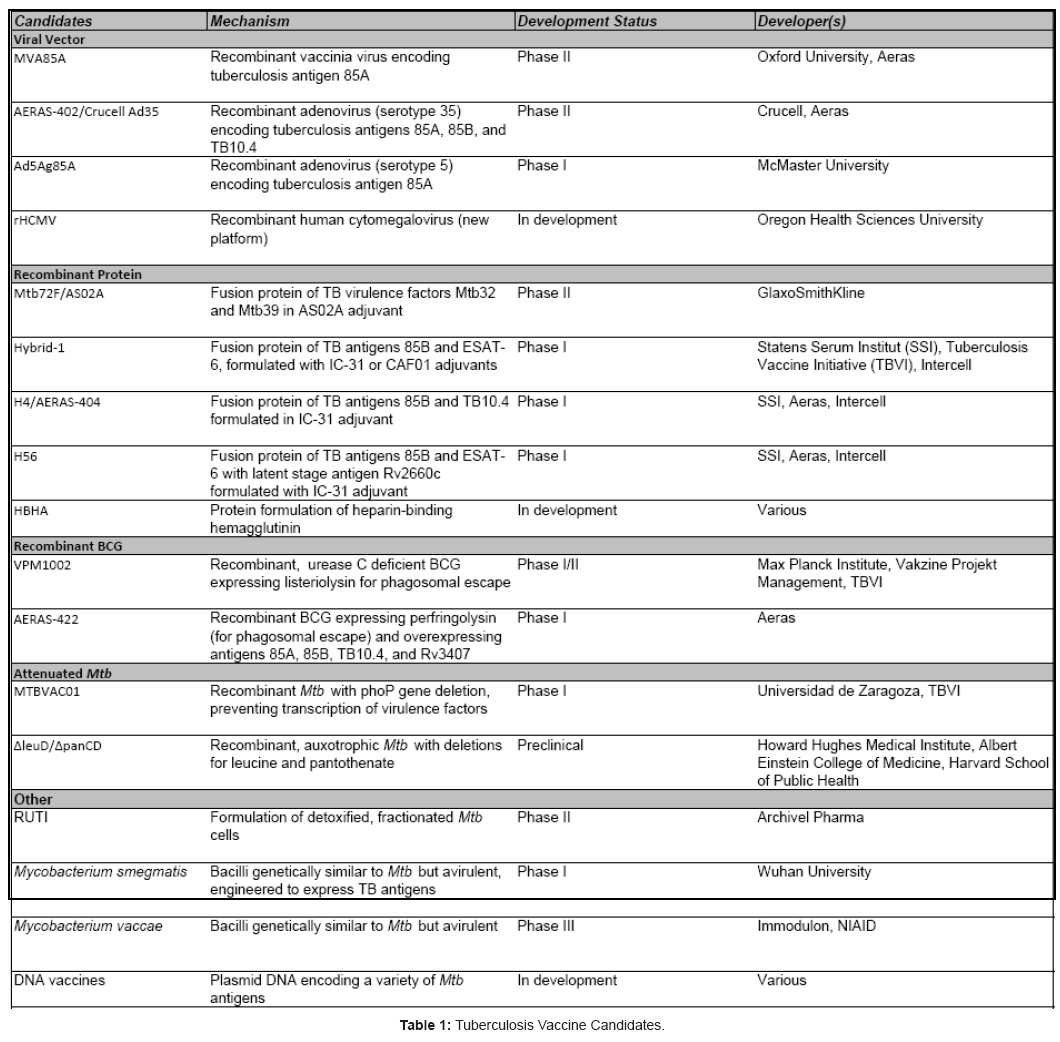
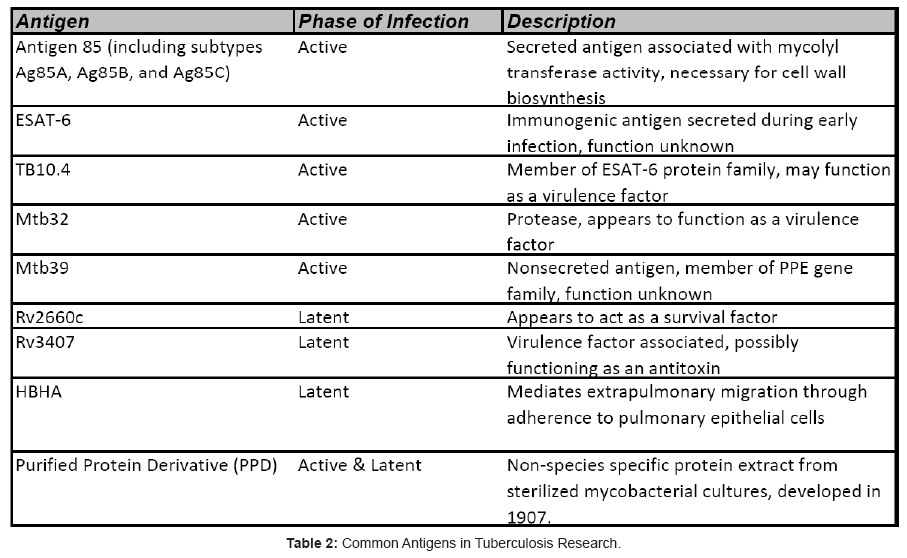
Vaccines in Development
Viral vector vaccines
Of the TB vaccines in development, vaccines based on a viral vector platform are among the most developed and characterized candidates. Using the machinery of the virus, these vaccines will deliver genetic information encoding an antigen to the host cells, where the antigen will be expressed as a protein. These viral vector candidates are targeted for a “heterologous prime-boost strategy” [31,32]. Simply put, the heterologous prime-boost strategy relies on priming the immune system with a different viral vector, a recombinant protein with adjuvant, or the readily-available BCG vaccine (or, in the future, a recombinant BCG replacement), followed at some point by a non-BCG viral booster vaccine. In this manner, the boosting vaccine is utilized to hone a robust response to Mtb antigens that may not have readily been presented to the host immune system by BCG and, specifically, to expand upon the CD8+ T cell response, and in some cases a CD4+ T cell response, in order to generate a stronger and more directed immune response to TB.
MVA85A
The antigen 85 complex, part of Mycobacterium tuberculosis, is made up of three proteins, commonly referred to as antigen 85A, 85B, and 85C [33]. These proteins are highly secreted, and have previously been shown to exhibit mycolyl transferase activity, which is vital for mycobacterial cell wall biosynthesis [34]. Furthermore, studies have revealed that these proteins are highly immunogenic [35].
Using this knowledge, researchers at Oxford University created MVA85A [36]. MVA85A is a recombinant modified vaccinia Ankara virus engineered to express the Mtb antigen 85A. Modified vaccinia Ankara, or MVA, is an attenuated strain of vaccinia virus that does not efficiently replicate in primate cells [37]. Furthermore, MVA was chosen as a platform because poxviruses have been shown to elucidate a strong T cell response when used as a boosting vaccine [38].
The first Phase I clinical trials of MVA85A were reported in 2004 [38]. These Phase I studies, conducted with healthy adult volunteers from the UK, indicated MVA85A was safe and effective. Of 31 test subjects, adverse effects were primarily limited to flu-like symptoms (headache, fever, nausea). Furthermore, 16 of 17 test subjects in the heterologous prime-boost group exhibited significant and sustained increases in antigen-specific T cells. These antigen-specific responses, measured using ex vivo interferon-γ ELISpot, were observed up to 24 weeks after vaccination. Supporting the results of these Phase I trials, additional data was published to characterize the polyfunctional nature of the T cell response to the MVA85A vaccine [39]. Using polychromatic flow cytometry, CD4+T cells from previously vaccinated humans were shown to produce multiple cytokines in response to stimulation with antigen 85A, as well as generate robust proliferation. These results were later confirmed in a study conducted in South Africa, again testing healthy adults [40].
Another critical Phase I study, published in 2009, tested MVA85A in patients with latent TB infection [41]. As with previous research, this study indicated the MVA85A vaccine was safe in patients infected with TB. Additionally, a significant increase in antigen-specific T cells was observed, suggesting the vaccine enhances immunogenicity even in patients that have already been exposed to Mtb. Similarly, a study published in 2010 suggests similar safety and immunogenicity in adolescents and children [42].
The scientific evidence gathered has advanced MVA85A into Phase II clinical trials. Recently, the first results of a Phase IIa clinical trial in healthy infants were reported [43]. These results have upheld the promising outlook established by the earlier trials: MVA85A appears to be very safe, and promotes robust immune responses. These early trials, though suggesting that MVA85A is a safe vaccine candidate that elicits a strong immune response, do leave one lingering question: does MVA85A promote protection from Mtbγ As the focus of research shifts to attempt to answer this question, MVA85A has now moved into Phase IIb “proof-of-concept” trials (based on ClinicalTrials.gov listings). Also, as the current heterologous prime-boost method that is employed relies on BCG for priming, there are limitations to using this vaccine if the patient is immunocompromised and has not been previously vaccinated with BCG. However, as MVA85A continues towards Phase III clinical trials, the consistent safety and immunogenicity data lends hope that this candidate may soon be among the first new preventative vaccines for TB since the introduction of BCG.
AERAS-402/Crucell Ad35
The AERAS-402/Crucell Ad35 vaccine is another viral vector vaccine, developed jointly by Aeras and Crucell. As with MVA85A, this vaccine candidate has been designed to be used in a heterologous prime-boost strategy [32]. Unlike MVA85A, however, this vaccine relies on a recombinant adenovirus, serotype 35 (Ad35) as the delivery vector. The adenoviral vector was chosen for a number of reasons. For one, this adenoviral vector has been shown to elicit CD8 responses and is replication deficient [44], potentially allowing use with immunocompromised patients. The Ad35 vector has also been shown to be safe in Phase I clinical studies [32]. Additionally, previous research has shown Ad35 to be effective when used as a boosting vector in prime-boost regimens [45]. Ad5, another adenoviral serotype, was considered, but research suggests that there is a high prevalence of neutralizing antibodies present in people residing in sub-Saharan Africa [46] that may lessen the value of an Ad5-based vaccine in that area.
With these factors in mind, Aeras and Crucell engineered an Ad35 vaccine that could express a fusion protein of three key Mtb antigens: antigen 85A (Ag85A), antigen 85B (Ag85B), and TB10.4 [32]. TB10.4 is believed to be a virulence factor in Mtb [47]. Preclinical studies in animal models showed promising results for the AERAS-402/Crucell Ad35 vaccine candidate, with strong polyfunctional T cell responses and Mtb protection observed when used as a boosting vaccine for an experimental recombinant BCG [48].
AERAS-402/Crucell Ad35 moved into Phase I clinical trials, which were first reported in 2010 [32]. This trial tested safety and immunogenicity of the vaccine in healthy adults in South Africa that were previously vaccinated with BCG. The trial characterized the safety of the AERAS-402/Crucell Ad35 vaccine with mainly mild-tomoderate adverse events. While the safety data was encouraging, the immunogenicity data proved even more promising. The trial data indicated that, in addition to a polyfunctional CD4+ T cell response, a strong CD8+ T cell response was also observed. The CD8+ T cell response may prove to be vital to the success of AERAS-402/Crucell Ad35, as most other TB vaccine candidates have reported only negligible CD8+ T cell responses [32].
According to ClinicalTrials.gov, AERAS-402/Crucell Ad35 has progressed to Phase II clinical trials, with one trial investigating the utility of the candidate in infants (ID# NCT01198366), and another trial investigating safety and efficacy in HIV+ subjects (ID# NCT01017536). The advancement of this vaccine candidate into Phase II trials represents promise for the future, but some limitations must first be addressed. As with MVA85A, there is no clear answer as to what immune responses are required for protection from Mtb. While the CD8+ T cell responses separate AERAS-402/Crucell Ad35 from MVA, protein/adjuvant, and recombinant BCG TB vaccine candidates, this impact of the CD8+ T cell response on TB protection remains uncertain. Another question that remains to be answered is the usefulness of the Ad35 vector. One important aspect of the Ad35-vectored vaccine is that Ad35 can be lyophilized and delivered as an inhalable powder, potentially eliciting an immune response that is localized in the lungs, which may be useful strategy for combating TB [49,50].
Ad5Ag85A
Developed by McMaster University, the Ad5Ag85A vaccine candidate is a viral vector vaccine based on adenovirus, serotype 5 [51]. As with Ad35 described above, initial testing of the Ad5 serotype suggested Ad5 is very safe for use in human vaccines [44]. The researchers at McMaster University utilize this vector to deliver Ag85A as the key antigen. First described in 2004, preclinical animal studies with this vaccine have shown promising results, characterized by high antigen-specific T cell responses [52-55]. One study even showed strong protection against Mtb in guinea pigs when the animals were boosted intranasally with Ad5Ag85A [56]. Other animal studies have focused on variations to the vaccine delivery method, showing enhanced immunity via intranasal vaccination [55,56]. The researchers have also engineered and tested a related vaccine that expresses both Ag85A and TB10.4 [57] that also showed strong immunogenicity.
According to ClinicalTrials.gov, one Phase I human clinical trial (ID# NCT00800670) is registered for the Ad5Ag85A vaccine, and the listing suggests the researchers intend to investigate this candidate as both a prime and a boost vaccine. Despite this, no results from human testing have yet been published. Without any published human data, it is difficult to describe the potential for this vaccine. This candidate undoubtedly faces many of the same questions as the previously outlined candidates. For one, will the expressed antigen not only induce immunogenicity, but lead to protection against Mtbγ Furthermore, will the vaccine maintain its safety profile as it is tested in different populations, including infants or HIV+ patientsγ Perhaps the biggest challenge, however, lies with the serotype of adenovirus employed by this candidate. As mentioned above, at least one study has suggested that up to 90% of the global population has neutralizing antibodies to Ad5 [45]. This is troubling because, if true, this statistic would indicate that only 10% of the global population may see a benefit from this vaccine candidate. At this point, however, it is impossible to determine the full implications of this statistic without some human clinical trial data to confirm or refute this pronouncement. Finally, the HIV STEP trial results raise concerns about using the Ad5 platform in areas with high seropositivity against Ad 5 and high incidences of HIV.
Recombinant protein vaccines
Recombinant protein vaccines have a long development history, including vaccine candidates against HIV [58], Lyme disease [59], and others. Rather than relying on a virus to deliver key antigenic DNA, recombinant protein vaccines seek to inject the fully formed antigen protein directly for presentation to the immune system [60].
In order to manifest a particular type of immune response to the recombinant protein, different adjuvants may be employed with the vaccine. For the tuberculosis vaccine candidates discussed, the popular adjuvants are ASO1, ASO2, IC31, and CAF01. ASO1 and ASO2 were both developed by GlaxoSmithKline (GSK). ASO1 was developed to deliver CD8+ T cell response, and is a liposome formulation containing QS21 (bark extract from Quillaja saponaria tree) and MPL (monophosphoryl lipid A from Gram-negative bacteria) with TLR4 agonistic effects [61]. AS02 was similarly formulated with QS21 and MPL, only instead in an oil/water emulsion [61]. IC31 is a proprietary adjuvant developed by Intercell. IC31 employs ODN1a (an oligodeoxynucleotide) and KLKL(5)KLK (a derivative of the cationic antimicrobial peptide) to act as a TLR9 agonist [62]. At least one study has suggested IC31 to be a potent activator of dendritic cells [62]. Finally, CAF01 is developed by Statens Serum Institut (SSI). The CAF01 adjuvant delivers a synthetic mycobacterial cord factor using DDA- (N,N’-dimethyl-N,N’-dioctadecylammonium) based liposomes [63]. CAF01 has been shown to trigger dendritic cell activation, and helps to induce Th1 and Th17 responses [62,64,65]. vaccine in a heterologous prime-boost strategy, other vaccines are being actively tested as a priming vaccine as well as a booster.
M72
M72 is a recombinant fusion protein in adjuvant manufactured by GSK. First reported in 2004, Mtb72F (the fusion protein of the M72 vaccine) is a 72-kDa recombinant protein fusing Mtb32 and Mtb39 [66]. Each of these proteins is secreted in culture filtrate, and each is suspected to be a virulence factor of Mtb. The initial animal studies with this vaccine tested the protein in combination with both AS02A and AS01B adjuvants, both of which are proprietary adjuvants developed by GSK. These tests showed moderate IFN-γ responses and minimal CD8+ T cell responses with the AS02A adjuvant, but markedly robust CD8+ T cell responses with the AS01B adjuvant.
Despite the results of this experiment, an additional study was conducted in guinea pigs examining co-vaccination with BCG and Mtb72F formulated with the AS02A adjuvant [67]. This experiment demonstrated that co-vaccination with Mtb72F significantly improved protection in guinea pigs following aerosol challenge with Mtb when compared to guinea pigs that were only vaccinated with BCG. Further preclinical studies supported the hypothesis that AS02A would be advantageous to AS01B due to safety and immunogenicity concerns [68], and the Mtb72F/AS02A vaccine was moved to Phase I clinical trials [66]. The first published results from a Phase I clinical trial testing Mtb72F/AS02A were reported in 2009 [69]. This trial assessed safety and immunogenicity in healthy adult volunteers. In regards to safety, the most common adverse events were local reactogenic symptoms. No major adverse events were reported. In terms of immunogenicity, significant increases in IL-2 and IFN-γ production were observed via ELISpot assays. Induction of CD4+ T cell activation markers was also observed via intracellular cytokine staining (ICS), but CD8+ T cell responses were negligible. A dose-escalation study reported similar results [70]. These results have prompted two Phase II clinical studies, one in infants (ID# NCT01098474) and one in HIV+ patients (ID# NCT01262976), according to ClinicalTrials.gov. GSK has also reformulated M72 in another proprietary adjuvant, AS01E (unpublished data).
Despite these successes, questions regarding the development of this vaccine candidate remain. One observed trend is the lack of CD8+ T cell responses. Though a robust CD4+ T cell response may be requisite for immunity to Mtb, CD8+ T cells will likely play a role in protection as the organism resides inside of macrophages. Further characterization is required to determine if the lack of a CD8+ T cell response will be detrimental to the success of this candidate.
Hybrid-1
Developed by SSI and the TB Vaccine Initiative (TBVI), Hybrid-1 or H1 is a fusion protein of Ag85B and ESAT-6 [71]. ESAT-6 is a 6 kDa secreted protein that has been well characterized as a potent T cell antigen [72]. In 2001, the first animal studies testing H1 were reported [71]. This early study suggested that H1 could elicit protective immunity in mice and guinea pigs observed up to 30 weeks post-vaccination. An additional preclinical study was conducted in nonhuman primates [73]. Interestingly, this study utilized H1 as the priming vaccine, and demonstrated that protection in nonhuman primates could be achieved without first priming with the BCG vaccine. This experiment also showed activation of both CD4+ and CD8+ T cells when combining H1 with two different adjuvants (AS02A and DDA/MPL). Interestingly, a partnership with Intercell examined the utility of the
H1 vaccine in the proprietary IC31 adjuvant [74]. This adjuvant was shown to help elicit Th1 CD4+ T cell responses. Preclinical testing with this combination showed high levels of CD4+ interferon-γ response in guinea pigs challenged with aerosol Mtb. These findings moved the H1/ IC-31 formulation into human clinical trials.
Two Phase I human clinical studies have been reported recently, with the first examining H1/IC-31 in healthy adult volunteers [75] , and the second in adult volunteers previously vaccinated with BCG or infected with Mtb [76]. In each case, the safety of the H1/IC-31 candidate was characterized by minor adverse events and no longterm negative effects. Immunogenicity data from each of these trials indicated a strong, antigen-specific T cell response characterized by increased IFN-γ production. These trials also showed that memory responses could be up to 32 months after vaccination. At least one additional Phase I study with this formulation is ongoing, according to ClinicalTrials.gov (ID# NCT01049282).
SSI is also investigating a formulation that combines H1 with the adjuvant CAF01 (cationic adjuvant formulation 01) [77]. The first preclinical study suggested that this alternative formulation of the H1 vaccine candidate was able to induce a robust polyfunctional CD4+ T cell response, also as a priming vaccine. Since that time, SSI has moved this alternative formulation, H1/CAF01, to Phase I human trials (ID# NCT00922363, ClinicalTrials.gov). Although the initial tests show potential, there are some shortcomings to this vaccine. One of the most prominent tools in diagnosing TB infection is the IFN-γ release assay (marketed as QuantiFERON-TB Gold), which relies on stimulation of human whole blood with ESAT-6 and other TB antigens [78]. If H1 were to be used in a widespread manner, the exposure to ESAT-6 from the H1 vaccine was feared to render the IFN-γ release assay less useful in diagnosing the BCG-vaccinated patients. The utility of this diagnostic tool is an important consideration in the development of the H1 vaccine candidate. However, the published trial data to this point have not shown ESAT-6 to interfere with the QuantiFERON-TB Gold test. Additionally, new diagnostic technologies for the detection of TB infection, such as the GeneXpert real-time PCR system [79], could eliminate this concern completely.
H4/Aeras-404
In anticipation of the diagnostic problem of using ESAT-6 as a vaccine component, SSI, Aeras, Intercell, and Sanofi Pasteur have developed the H4/AERAS-404 vaccine [80,81]. This vaccine employs a fusion protein that combines TB10.4, rather than ESAT-6, with antigen 85B, as experiments indicated TB10.4 may be a powerful antigen for TB research [82]. The first preclinical study, reported in 2005, indicated similar immunogenicity results to H1 when used as a priming vaccine, with the added advantage of maintaining diagnostic sensitivity to ESAT-6 [80]. A second preclinical study examined the utility of H4/ AERAS-404, adjuvanted with IC-31, as a boosting vaccine following BCG vaccination [81]. This study demonstrated superior protection to BCG, with reduced lung involvement in guinea pigs challenged with Mtb. Safety data from this study indicated H4/AERAS-404 should be well tolerated, and supported beginning Phase I clinical trials. One review [83] indicates Phase I clinical trials are ongoing with H4/ AERAS-404, though published results are not yet available. As such, it is difficult to conclude if the safety and immunogenicity demonstrated in the preclinical studies has carried over to human trials.
H56
Each of the vaccines described to this point rely heavily on presenting antigens that are present in the early stages of infection. The researchers at SSI have hypothesized that later-stage antigens may be important in preventing latent TB infection [84]. Using this strategy, SSI has developed H56, which is a fusion protein of early stage antigens 85B and ESAT-6, as well as the latent stage antigen Rv2660c. Previous research has demonstrated Rv2660c can elicit an enhanced CD4+ T cell response in patients with latent TB infection [85]. Applying this knowledge, SSI specifically sought to test the application of the H56 vaccine as a multistage vaccine, meaning that this vaccine could be used as both a preventative and a therapeutic vaccine [84]. The results of the first published animal studies indicated that H56 conferred protective immunity in three different mouse models, as well as reduced bacterial load in latent TB mouse models. Based on these results, collaboration between SSI, Aeras, and Intercell has moved H56, formulated in IC31 adjuvant, toward Phase I clinical trials [86]. Although the developmental strategy for this vaccine is unique and offers an attractive multistage vaccine candidate, its utility in humans is yet to be determined.
Recombinant BCG
Given the proven safety record of BCG, it is not surprising that several groups are looking to utilize the existing vaccine as a platform for a newer, improved BCG vaccine. To improve vaccine efficacy, a number of theories have been generated. As mentioned previously, interference with the phagosome maturation process has led to the understanding that BCG, unlike Mycobacterium tuberculosis, does not escape the endosome of an infected macrophage [87]. This has led to the theory that significant antigens from the BCG vaccine are not being properly presented, perhaps leading to defective protection. In hopes of altering the BCG presentation pathway, researchers have taken the novel approach of adding genes encoding bacterial toxins capable of lysing the phagosomal membrane to allow BCG to escape to the cytosol (additional information below). Other research has strongly indicated that a potent Th1 CD4+ T cell response, leading to CD8+ T cell activation, may be a key factor for developing immunity to TB [88-90]. For the vaccine candidates described below, researchers have genetically manipulated BCG in hopes to enhance antigen presentation compared to the original vaccine.
VPM1002
One of the first recombinant BCG (rBCG) vaccine candidates to reach human clinical trials, VPM1002 is being developed through a collaboration of the Max Planck Institute, Vakzine Projekt Management GmbH, and the Tuberculosis Vaccine Initiative (TBVI) [83]. First described in a study published in 2005, VPM1002 is based on the BCG Prague strain [91]. Researchers constructed a recombinant version of this BCG to express listeriolysin, a toxin secreted by Listeria monocytogenes that is known to lyse phagosomes and allows L. monocytogenes to escape into the cytosol [92]. Furthermore, VPM1002 was constructed with a urease C (ureC) deficiency. Urease C functions in mycobacteria to neutralize the pH of the phagosome to improve mycobacterial survival [93]. As listeriolysin functions best at a low pH [94], the ureC deficiency was necessary for VPM1002 to function as designed. Reported preclinical studies indicated VPM1002 performed better than BCG in protecting mice from Mtb challenge. Specifically, the listeriolysin from VPM1002 was shown to promote translocation from the phagosome and promote apoptosis of infected macrophages. The developers have surmised that the phagosome escape mechanism augments T cell immunity by allowing antigens to gain access to the cytosolic antigen processing machinery for presentation to CD8+ T cells. A phase Ib clinical study is currently underway (ClinicalTrials.gov, ID# NCT01113281. A previous review indicated a phase II trial in infants is planned for VPM1002 [83]. Though the phagosome escape mechanism is a significant advancement for rBCG candidates, it remains to be seen as to whether this mechanism will make BCG more efficacious. Furthermore, the inclusion of a gene encoding for a bacterial toxin (listeriolysin) may cause additional regulatory scrutiny for safety of this candidate. However, no hemolysis or other cytotoxicity was observed during preclinical animal studies.
Aeras-422
For years, Aeras has been developing a number of rBCG candidates [48,95,96]. In 2010, Aeras advanced one of its rBCG candidates, AERAS-422, to Phase I clinical trials [86]. AERAS-422 is a recombinant BCG constructed to overexpress antigens 85A, 85B, and Rv3407. Similar to Rv2660c in the H56 vaccine candidate, Rv3407 was chosen due to previous studies that indicate it is an antigen associated with latent TB infection [97]. In addition to the overexpressed antigens, AERAS-422 was also engineered to produce perfringolysin O and lack ureC expression [86,96]. Perfringolysin O is a cytolysin from Clostridium perfringens [98]. Much like lysteriolysin from VPM1002, perfringolysin O is employed to enable the rBCG to escape the phagosome such that its antigens can be presented via the MHC class I pathway. Aeras has previously reported results from preclinical studies from AERAS-422 precursors designated AERAS-401 and AFRO-1 [96]. The results of these studies indicated that the AERAS-422 precursors prolonged survival of mice challenged with Mtb, and was safe even in immunocompromised (SCID) mice. The first phase I clinical study is underway (ClinicalTrials.gov, ID# NCT01340820), with no published human data yet available. The phagosome escape mechanism, coupled with the overexpressed antigen system, presents a strong theoretical candidate for further development. As with VPM1002, the inclusion of bacterial toxin gene products into this candidate may cause regulatory concerns. Once again, however, hemolysis or cytotoxicity was not observed in animal studies, including the SCID mouse study. Additional safety data is required in order to determine if this candidate will prove safer than the parent BCG
Attenuated Mtb
One of the disadvantages to each of the previously mentioned candidates is that the entire repertoire presents (or overexpresses) a relatively limited library of possible antigens. Though the preliminary data generated from each of those candidates suggests generation of immunogenicity, it remains undetermined as to which, if any, of the antigens utilized will generate protective immunity to Mtb. With this in mind, several groups are developing an attenuated Mtb strain for use in vaccination. The concept behind these candidates is to genetically modify Mtb such that it cannot replicate or cause pathogenesis. In this way, vaccination with the mutant Mtb can present the full range of antigens without the threat of disease. Furthermore, as these candidates are replication deficient, the concern for use in immunocompromised patients is theoretically muted. Importantly, prior exposure to Mtb does not ensure protection from subsequent exposure, raising concerns about the feasibility of attenuated Mtb as an effective vaccine platform. However, this lack of a protective response may be attributed to virulence factors within the bacteria. Continued research is necessary to address these important questions. Despite these concerns, attenuated Mtb has performed well in animal models (below) suggesting this may be a viable platform.
MTBVAC01
MTBVAC01, an attenuated Mtb vaccine based on the SO2 strain, was developed by the Carlos Martin lab at Universidad de Zaragoza in Spain [99]. The SO2 strain contains a mutation that prevents the Mtb bacilli from expressing the phoP gene, which is a component necessary for the transcription of key virulence factors [100]. Among other virulence factors, research has suggested that phoP is necessary for the secretion of ESAT-6 [101]. Using this information, the Martin lab tested SO2 for safety and efficacy in mice and guinea pigs [99]. These experiments showed that subcutaneous vaccination with SO2 caused greater protective immunity in guinea pigs than BCG following aerosol challenge with wild-type Mtb. The researchers also concluded that the SO2 strain of Mtb was more attenuated than BCG as observed in SCID mice. Coupled with previous results suggesting the SO2 bacilli are replication-deficient in macrophages [100], studies advanced to nonhuman primates. The first results from nonhuman primate studies were published in 2009 [102]. Using Rhesus macaques, this study compared BCG, MVA85A coupled as a boost following BCG prime, and a single-dose of SO2. The results suggest that both SO2 and the heterologous prime boost BCG/MVA85A strategy were welltolerated and superior to BCG alone in terms of immunogenicity and efficacy. Using SO2 as a precursor, MTBVAC01 was created by also incorporating a fadD26 deletion (unpublished data). Previous studies have indicated that fadD26 is an important virulence factor in Mtb [103]. Encouraged by these results, TBVI has recently announced that, in collaboration with the Martin lab, it is advancing MTBVAC01 to phase I human clinical trials [104].
Several challenges remain in the development of MTBVAC01, however. Despite its replication deficiency in macrophages, another study showed that the SO2 strain could replicate in pulmonary fibroblasts [105]. Additional safety studies in animals were performed to ensure that the attenuation of the SO2 mutant strain is adequate [106]. However, despite all of this, governmental regulators may be hesitant to approve an attenuated Mtb vaccine out of fear of genetic reversion.
Another question remaining in the development of MTBVAC01 is its ability to prevent an Mtb infection with an Mtb based vaccine. Previous epidemiological studies, including one performed in South Africa [107], indicate that previous infection and treatment for Mtb did not protect against subsequent reinfection. The South Africa study suggested the protection was particularly weak against MDR- and XDR-TB strains. Whether or not MTBVAC01 will protect against other strains of Mtb remains an open question.
ΔleuD/ΔpanCD
In an effort to develop an attenuated Mtb vaccine candidate that contains more than a single genetic mutation but maintains efficacy, a collaborative effort undertaken by faculty at several academic institutions has yielded a ΔleuD/ΔpanCD attenuated Mtb strain [108]. This strain is auxotrophic in that it cannot manufacture leucine or pantothenate due to two separate genetic deletions. The initial tests demonstrated protection in the guinea pig aerosol challenge model, as well as full attenuation in SCID mice. Recent results show that the ΔleuD/ΔpanCD strain provided better protection in rhesus macaques following aerosol challenge with Mtb than BCG [109]. The same study utilized SIV-infected macaques as an immunodeficiency model to demonstrate full attenuation of the ΔleuD/ΔpanCD strain. Although the researchers appear interested in moving this candidate forward into human clinical trials, it does not yet appear that this milestone has been reached for this candidate. As with MTBVAC01, it remains to be seen as to whether the genetic mutations in this strain cause sufficient attenuation for expansive use in humans, or as to whether or not an Mtb based vaccine can prevent infection from other Mtb strains.
Other Vaccines in Development
RUTI
Developed by Pere Juan Cardona and Archivel Pharma, RUTI is a formulation of detoxified, fractionated Mtb cells [110,111]. An initial preclinical study examined the utility of RUTI as a complimentary immunotherapy to chemotherapeutic agents used for treating TB infection [111]. This study suggested that RUTI enhanced both the Th1/ Th2 responses in two different mouse TB models. An additional study showed that RUTI decreased the bacterial load in both mice and guinea pigs infected with Mtb compared to post-infection immunization with BCG [112]. Positioned somewhat uniquely as a post-infection immunotherapy, RUTI was advanced to human clinical trials. The first phase I clinical study was performed with healthy adult volunteers in order to assess the safety profile of RUTI [110]. RUTI demonstrated dose-dependent local reactions, but was otherwise well-tolerated. RUTI was also shown to generate T cell responses specific to purified protein derivative (PPD) and other Mtb antigens. A phase II study in both HIV+ and HIV- patients being treated for latent TB infection was recently completed (ClinicalTrails.gov, ID# NCT01136161), with results yet to be published. An additional preclinical study sought to examine the utility of RUTI as a preventative vaccine [113]. This study indicated that RUTI, administered prior to aerosol challenge of Mtb, did reduce the bacterial load in both the lung and spleen in mice. However, BCG was more effective at reducing bacterial load in the spleen than RUTI. RUTI also improved the survival of guinea pigs when used as a prophylactic vaccine. The authors suggested future experiments may look to further characterize RUTI as a preventative vaccine. RUTI presents an interesting vaccine candidate. On one hand, it is one of the few candidates to be used as an immunotherapy following infection and show immunogenicity in this role. Furthermore, as fractionated Mtb, RUTI will employ an array of antigens beyond those being explored by other vaccine candidates. However, the utility as a preventative vaccine may be limited. In addition to results that suggest BCG may be superior in some regards [113], the exposure to an amalgam of Mtb antigens may invalidate a number of diagnostic tools if RUTI is used in uninfected patients.
Mycobacterium smegmatis
Mycobacterium smegmatis is a bacilli from the same genus as Mycobacterium tuberculosis [114]. Unlike its Mtb cousin, however, M. smegmatis is avirulent in humans, providing a genetically similar vector that may be an attractive vaccine candidate. However, M. smegmatis on its own does not appear to generate an anti-tuberculosis immune responses due to poor presentation of immunogenic Mtb antigens [115]. To compensate for its immunogenic shortcomings, several groups are examining recombinant versions of Mycobacterium smegmatis that overexpress a variety of key Mtb antigens [115,116] or genes for enhanced immunity [117]. These published results indicated safety and immunogenicity in preclinical animal models. Wuhan University in China has communicated that it is advancing a Mycobacterium smegmatis vaccine candidate to phase I human clinical trials [118].
Mycobacterium vaccae
Mycobacterium vaccae is another bacillus of the Mycobacterium genus. Given its genetic similarity to Mtb, investigators have been researching the use of M. vaccae as a tuberculosis immunotherapeutic agent since at least 1985, when one study suggested M. vaccae could be useful in improving BCG immune responses [119]. Since that time, a number of studies have continued to indicate that heat-inactivated M. vaccae improves immunity to Mtb when used in coordination with antibiotics [120-127]. Given its long development history, it is not surprising to learn that Immodulon and NIAID have recently completed a phase III clinical study utilizing a version of M. vaccae as a preventative vaccine [127]. This trial studied the utility of M. vaccae in preventing tuberculosis in HIV+ individuals that have been previously vaccinated with BCG. The results of this trial indicated M. vaccae was safe for use in immunocompromised individuals, and that M. vaccae induced improved protection against tuberculosis. Despite this success, a recent review indicated reformulation of the vaccine was pending [60], and no further trials with this particular candidate have been reported to ClinicalTrials.gov.
Future Platforms
rHCMV
One of the newest vaccine platforms to be described is that of recombinant human cytomegalovirus (rHCMV) [128]. Human cytomegalovirus is a member of the herpesvirus family, with nearly ubiquitous infection in all adult humans [129]. Interestingly, prior infection with CMV does not prevent re-infection such that it is possible to give homologous rHCMV boosts in a vaccine regimen. Due to its persistent nature as a β-herpesvirus, HCMV is unique in its ability to induce high frequency effector memory T cell responses that persist for significant periods of time. These effector populations have been shown to traffic to peripheral tissues, including the lung, at high levels. Together, these factors make rHCMV an attractive viral vaccine platform. Recently, one group has published promising results in priming a protective response in rhesus macaques against SIV [128]. Using the rHCMV vector to present SIV antigens, a robust effector memory T cell response was observed. Further study results were published, suggesting the rHCMV vector provided protective immunity in 12 of 13 challenged animals [130]. The results have obvious implications for the HIV vaccine field. However, the impact of this data reaches beyond HIV and research is currently underway to assess this novel vector for other diseases, including TB. It should also be noted, however, that due to the species specificity of CMV viruses, the rhesus CMV used in the previous study is only a proof of concept study. Due to the persistent nature of the virus, and the potential adverse events associated with CMV infection (i.e. congenital birth defects), the development of an attenuated human CMV vector will take several years.
HBHA
Heparin-binding hemagluttinin, or HBHA, is a new antigenic TB vaccine target that is undergoing preclinical testing for immunogenicity. HBHA is a protein associated with Mtb migration from the lung to extrapulmonary tissues [131], and is generally described as a latent phase antigen [132].
HBHA has been shown to induce strong immune responses and protection in animal models [133,134]. However, the effectiveness of HBHA has been linked to post-translational methylation [135], which is difficult to control for during vaccine development. Additional research is required to characterize the effect of this antigen as it relates to the challenges of its utilization.
DNA vaccines
DNA vaccines are another platform currently under investigation in preclinical animal models. Simply put, these vaccines inject plasmid DNA encoding TB antigen(s) into a patient. Following uptake by a host cell, the plasmid is expressed and the resulting protein antigen is produced using host cell machinery.
A number of DNA vaccine candidates utilizing a variety of antigenic targets are under development and have been tested in animal models [136,137]. Furthermore, much of the literature available has suggested these vaccines are safe and immunogenic. However, to date, none of these TB DNA vaccine candidates have been advanced into clinical trials. However, DNA vaccines have advanced further and performed well in the HIV field where very high response rates have been observed in phase I clinical trials in which electroporation devices are utilized. The combination of DNA with electroporation greatly increases the immunogenicity of DNA vaccines and the addition of cytokine-encoding plasmids, such as IL-12p70, allows for enhanced Th1 immunity. Early data also suggest that DNA vaccines with electroporation are safe and well-tolerated, emphasizing the feasibility of this vaccine platform in other disease models such as tuberculosis.
The Challenges Ahead
This review has outlined a significant number of tuberculosis vaccine candidates seeking to replace or enhance the 90-year old BCG vaccine. Though this robust pipeline offers hope that an effective tuberculosis prevention measure may be on the horizon, there are still several obstacles to achieving this goal. The primary challenges that must be overcome are funding, development of clinical trial sites, and establishing correlates of protection.
As with many fields of disease research, the current economic climate has placed an increasing hardship on TB vaccine developers. TB is not as problematic in the United States and other developed countries, and is subsequently not viewed as a looming public health threat by the general public. With only a small market in the U.S. for a new TB vaccine, pharmaceutical companies are not contributing much funding or research to the development of a new TB vaccine. Therefore, TB vaccine developers rely primarily on grants from governments and charitable organizations. The worldwide economic distress has led to decreased funding from these sources, just as a number of candidates are reaching the transition from phase II to phase III clinical trials. The synergistic effect of these conditions is causing hardships for many developers, causing them to seek new and diverse funding opportunities and thereby removing emphasis on science and medical research.
Another challenge is the development of clinical sites. Tuberculosis is endemic in a large number of developing countries. Many of these countries lack the established infrastructure required to operate a large-scale clinical trial with success. To remedy this, funds and efforts must be directed to either establishing new clinical trial sites or repurposing previously utilized sites. As with the funding challenge described above, these necessary activities divert time and money away from scientific and medical research. Collaborative use of vaccine trial sites may also reduce the costs associated with setting up and running large scale human trials. As funds become tighter, this strategy may become more important.
Correlates of protection also remain a nagging problem in the TB vaccine field. Specifically, researchers are struggling to connect which antigen-specific cytokine responses, or combinations of responses, are necessary for protection and in what magnitude. Protection may be tied to a specific protein or group of proteins or could be defined by a particular type of immune response. Currently, the most common vaccine responses measured are IFN-γ, IL-2, and TNF following peptide stimulation ex vivo. However, these responses may not be predictive of vaccine efficacy. In fact, a recent phase III study examining M. vaccae indicated that the immune responses measured were not predictive of Mtb protection [120]. Other facets of the immune response may be required, such as Th2, Th17, or even regulatory T cells to mediate inflammation in the lungs. The ability to define a correlate is even more difficult in light of the possibility that the correlate may not be tied to protein antigens. Lipids and glycolipids may contribute significantly to protection. Furthermore, the innate immune system may also be critical, particularly neutrophils and NK cells. Understanding how vaccines can impact innate immunity upon exposure to virulent TB is an area that is not well-understood. These questions must be addressed in order to characterize a new vaccine and potentially to identify a correlate. Meanwhile, the only measure of a vaccine candidate’s efficacy remains expensive, long-term prospective studies.
Compounding the correlates of protection problem are gaps in our knowledge of the immune system, particularly in newly described T cell subsets. One example is generation of T cell memory. Central and effector memory T cells are known to exist in CD8+ cytotoxic T cells, but evidence suggests that similar subsets exist under the CD4+ lineage [138]. Which of these memory subsets may be significant for protective immunity against tuberculosis is poorly defined. CD4+ Th17 cells have also been described recently [139], yet exactly how they function and their role in the immune system, and particularly in tuberculosis immunity, remains largely uncharacterized. Also troubling is how little is understood about tuberculosis antigens. Many of the common antigens utilized in the vaccine candidates reviewed are not fully characterized. Though these antigens have been shown to be immunogenic, a better understanding is required for the field to advance. Mycobacterium tuberculosis is also known to contain a number of lipid antigens in its cell wall [140], yet the role of lipid-specific T cells, particularly NK T cells, in protection against tuberculosis is largely unknown .
Another hurdle in the development of these vaccine candidates is the availability of animal models. Currently, there is no consensus animal model for TB research. Seemingly, each experiment in TB vaccine development is performed using a different model: some with mice, others with nonhuman primates, and still others with guinea pigs. Each species has advantages and weaknesses, and different strains within a species can provide different results. At this moment, there is not one particular animal model that effectively mimics TB infection in humans. Until such a model is developed, researchers will continue to rely upon numerous animal models, creating confusion and possibly inconsistent data.
Mycobacterium tuberculosis remains a global threat to human health despite its long history. After a prolonged reliance on BCG and antibiotics, new efforts are being undertaken to develop new, safe, and efficacious TB vaccines. These efforts have yielded a number of promising candidates, but also several intimidating challenges. Despite these challenges, a new preventative TB vaccine remains an important global health goal, obtainable perhaps by the end of the decade.
References
- Hershkovitz I, Donoghue HD, Minnikin DE, Besra GS, Lee OY, et al. (2008) Detection and molecular characterization of 9,000-year-old Mycobacterium tuberculosis from a Neolithic settlement in the Eastern Mediterranean. PloS one 3: e3426.
- Fisher L (1971) Rifampin--new and potent drug for TB treatment. Bull Natl Tuberc Respir Dis Assoc 57: 11-12.
- (1990) Outbreak of multidrug-resistant tuberculosis--Texas, California, and Pennsylvania MMWR Morb Mortal Wkly Rep 39: 369-72.
- (2006) Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs--worldwide, 2000-2004. MMWR Morb Mortal Wkly Rep 55: 301-305.
- Prevention CfDCa (2000) Biological and chemical terrorism: strategic plan for preparedness and response. Morb Mortal Wkly Rep 49: 1-14.
- (2003) Treatment of tuberculosis. MMWR Morb Mortal Wkly Rep Centers for Disease Control 52: 1-77.
- Oettinger T, Jorgensen M, Ladefoged A, Haslov K, Andersen P (1999) Development of the Mycobacterium bovis BCG vaccine: review of the historical and biochemical evidence for a genealogical tree. Tubercle and lung disease 79: 243-250.
- Trunz BB, Fine P, Dye C (2006) Effect of BCG vaccination on childhood tuberculous meningitis and miliary tuberculosis worldwide: a meta-analysis and assessment of cost-effectiveness. Lancet 367: 1173-1180.
- Colditz GA, Brewer TF, Berkey CS, Wilson ME, Burdick E, et al. (1994) Efficacy of BCG vaccine in the prevention of tuberculosis. Meta-analysis of the published literature. JAMA 271: 698-702.
- Fine PE (1995) Variation in protection by BCG: implications of and for heterologous immunity. Lancet 346: 1339-1345.
- Packe GE, Innes JA (1988) Protective effect of BCG vaccination in infant Asians: a case-control study. Arch Dis Child 63: 277-281.
- King L (2001) Minimising the risk of hospital transmission of pulmonary TB. Nurs Stand 16: 45-52.
- Ninane J, Grymonprez A, Burtonboy G, Francois A, Cornu G (1988) Disseminated BCG in HIV infection. Arch Dis Child 63: 1268-1269.
- Boudes P, Sobel A, Deforges L, Leblic E (1989) Disseminated Mycobacterium bovis infection from BCG vaccination and HIV infection. JAMA 262: 2386.
- Sterne JA, Rodrigues LC, Guedes IN (1998) Does the efficacy of BCG decline with time since vaccination? Tubercle and lung disease 2: 200-207.
- Rodrigues LC, Smith PG (1990) Tuberculosis in developing countries and methods for its control. Trans R Soc Trop Med Hyg 84: 739-744.
- Stanford JL, Shield MJ, Rook GA (1981) How environmental mycobacteria may predetermine the protective efficacy of BCG. Tubercle 62: 55-62.
- Turner J, Rhoades ER, Keen M, Belisle JT, Frank AA, et al. (2000) Effective preexposure tuberculosis vaccines fail to protect when they are given in an immunotherapeutic mode. Infect Immun 68: 1706-1709.
- Hasan Z, Schlax C, Kuhn L, Lefkovits I, Young D, et al. (1997) Isolation and characterization of the mycobacterial phagosome: segregation from the endosomal/lysosomal pathway. Molecular microbiology 24: 545-553.
- Ellner JJ (1997) Review: the immune response in human tuberculosis--implications for tuberculosis control. J Infect Dis 176: 1351-1359.
- Scanga CA, Mohan VP, Yu K, Joseph H, Tanaka K, et al. (2000) Depletion of CD4(+) T cells causes reactivation of murine persistent tuberculosis despite continued expression of interferon gamma and nitric oxide synthase 2. JEM 192: 347-358.
- van Pinxteren LA, Cassidy JP, Smedegaard BH, Agger EM, Andersen P (2000) Control of latent Mycobacterium tuberculosis infection is dependent on CD8 T cells. European journal of immunology 30: 3689-3698.
- Chen CY, Huang D, Wang RC, Shen L, Zeng G, et al. (2009) A critical role for CD8 T cells in a nonhuman primate model of tuberculosis. PLoS pathogens 5: e1000392.
- Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, et al. (1993) Disseminated tuberculosis in interferon gamma gene-disrupted mice. JEM 178: 2243-2247.
- Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, et al. (1993) An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. JEM 178: 2249-2254.
- Bean AG, Roach DR, Briscoe H, France MP, Korner H, et al. (1999) Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. Journal of Immunology 162: 3504-3511.
- Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, et al. (1995) Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2: 561-572.
- Mohan VP, Scanga CA, Yu K, Scott HM, Tanaka KE, et al. (2001) Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: possible role for limiting pathology. Infect Immun 69: 1847-1855.
- Day CL, Abrahams DA, Lerumo L, Janse van Rensburg E, Stone L, et al. (2011) Functional capacity of Mycobacterium tuberculosis-specific T cell responses in humans is associated with mycobacterial load. jimmunol 187: 2222-2232.
- Wilkinson KA, Wilkinson RJ (2010) Polyfunctional T cells in human tuberculosis. Eur J Immunol 40: 2139-2142.
- Nicol MP, Grobler LA (2010) MVA-85A, a novel candidate booster vaccine for the prevention of tuberculosis in children and adults. Curr Opin Mol Ther 12: 124-134.
- Abel B, Tameris M, Mansoor N, Gelderbloem S, Hughes J, et al. (2010) The novel tuberculosis vaccine, AERAS-402, induces robust and polyfunctional CD4+ and CD8+ T cells in adults. Am J Respir Crit Care Med 181: 1407-1417.
- Harth G, Horwitz MA, Tabatadze D, Zamecnik PC (2002) Targeting the Mycobacterium tuberculosis 30/32-kDa mycolyl transferase complex as a therapeutic strategy against tuberculosis: Proof of principle by using antisense technology. PNAS 99: 15614-15619.
- Belisle JT, Vissa VD, Sievert T, Takayama K, Brennan PJ, et al. (1997) Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 276: 1420-1422.
- Ulmer JB, Liu MA, Montgomery DL, Yawman AM, Deck RR, et al. (1997) Expression and immunogenicity of Mycobacterium tuberculosis antigen 85 by DNA vaccination. Vaccine 15: 792-794.
- McShane H, Behboudi S, Goonetilleke N, Brookes R, Hill AV (2002) Protective immunity against Mycobacterium tuberculosis induced by dendritic cells pulsed with both CD8(+)- and CD4(+)-T-cell epitopes from antigen 85A. Infect Immun 70: 1623-1626.
- Sutter G, Staib C (2003) Vaccinia vectors as candidate vaccines: the development of modified vaccinia virus Ankara for antigen delivery. Curr Drug Targets Infect Disord 3: 263-271.
- McShane H, Pathan AA, Sander CR, Keating SM, Gilbert SC, et al. (2004) Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nature medicine 10: 1240-1244.
- Beveridge NE, Price DA, Casazza JP, Pathan AA, Sander CR, et al. (2007) Immunisation with BCG and recombinant MVA85A induces long-lasting, polyfunctional Mycobacterium tuberculosis-specific CD4+ memory T lymphocyte populations. Eur J Immunol 37: 3089-3100.
- Hawkridge T, Scriba TJ, Gelderbloem S, Smit E, Tameris M, et al. (2008) Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in healthy adults in South Africa. J Infect Dis 198: 544-552.
- Sander CR, Pathan AA, Beveridge NE, Poulton I, Minassian A, et al. (2009) Safety and immunogenicity of a new tuberculosis vaccine, MVA85A, in Mycobacterium tuberculosis-infected individuals. Am J Respir Crit Care Med 179: 724-733.
- Scriba TJ, Tameris M, Mansoor N, Smit E, van der Merwe L, et al. (2010) Modified vaccinia Ankara-expressing Ag85A, a novel tuberculosis vaccine, is safe in adolescents and children, and induces polyfunctional CD4+ T cells. Eur J Immunol 40: 279-290.
- Scriba TJ, Tameris M, Mansoor N, Smit E, van der Merwe L, et al. (2011) Dose-Finding Study of the Novel Tuberculosis Vaccine, MVA85A, in Healthy BCG-Vaccinated Infants. J Infect Dis 203: 1832-1843.
- Barnett BG, Crews CJ, Douglas JT (2002) Targeted adenoviral vectors. Biochimica et biophysica acta 1575: 1-14.
- Radosevic K, Rodriguez A, Lemckert A, Goudsmit J (2009) Heterologous prime-boost vaccinations for poverty-related diseases: advantages and future prospects. Expert Rev Vaccines 8: 577-592.
- Kostense S, Koudstaal W, Sprangers M, Weverling GJ, Penders G, et al. (2004) Adenovirus types 5 and 35 seroprevalence in AIDS risk groups supports type 35 as a vaccine vector. AIDS 18: 1213-1216.
- Kato-Maeda M, Rhee JT, Gingeras TR, Salamon H, Drenkow J, et al. (2001) Comparing genomes within the species Mycobacterium tuberculosis. Genome Res 11: 547-554.
- Magalhaes I, Sizemore DR, Ahmed RK, Mueller S, Wehlin L, et al. (2008) rBCG induces strong antigen-specific T cell responses in rhesus macaques in a prime-boost setting with an adenovirus 35 tuberculosis vaccine vector. PloS one 3: e3790.
- Geisbert TW, Bailey M, Hensley L, Asiedu C, Geisbert J, et al. (2011) Recombinant adenovirus serotype 26 (Ad26) and Ad35 vaccine vectors bypass immunity to Ad5 and protect nonhuman primates against ebolavirus challenge. J Virol 85: 4222-4233.
- Hokey DA, Misra A (2011) Aerosol vaccines for tuberculosis: a fine line between protection and pathology. Tuberculosis 91: 82-85.
- Wang J, Thorson L, Stokes RW, Santosuosso M, Huygen K, et al. (2004) Single mucosal, but not parenteral, immunization with recombinant adenoviral-based vaccine provides potent protection from pulmonary tuberculosis. Journal of immunology 173: 6357-6365.
- Roediger EK, Kugathasan K, Zhang X, Lichty BD, Xing Z (2008) Heterologous boosting of recombinant adenoviral prime immunization with a novel vesicular stomatitis virus-vectored tuberculosis vaccine. Mol Ther 16: 1161-1169.
- Santosuosso M, McCormick S, Zhang X, Zganiacz A, Xing Z (2006) Intranasal boosting with an adenovirus-vectored vaccine markedly enhances protection by parenteral Mycobacterium bovis BCG immunization against pulmonary tuberculosis. Infect Immun 74: 4634-4643.
- Malowany JI, McCormick S, Santosuosso M, Zhang X, Aoki N, et al. (2006) Development of cell-based tuberculosis vaccines: genetically modified dendritic cell vaccine is a much more potent activator of CD4 and CD8 T cells than peptide- or protein-loaded counterparts. Mol Ther 13: 766-765.
- Santosuosso M, Zhang X, McCormick S, Wang J, Hitt M, et al. (2005) Mechanisms of mucosal and parenteral tuberculosis vaccinations: adenoviral-based mucosal immunization preferentially elicits sustained accumulation of immune protective CD4 and CD8 T cells within the airway lumen. Journal of immunology 174: 7986-94.
- Xing Z, McFarland CT, Sallenave JM, Izzo A, Wang J, et al. (2009) Intranasal mucosal boosting with an adenovirus-vectored vaccine markedly enhances the protection of BCG-primed guinea pigs against pulmonary tuberculosis. PloS one 4: e5856.
- Mu J, Jeyanathan M, Small CL, Zhang X, Roediger E, et al. (2009) Immunization with a bivalent adenovirus-vectored tuberculosis vaccine provides markedly improved protection over its monovalent counterpart against pulmonary tuberculosis. Mol Ther 17: 1093-1100.
- Belshe RB, Clements ML, Dolin R, Graham BS, McElrath J, et al. (1993) Safety and immunogenicity of a fully glycosylated recombinant gp160 human immunodeficiency virus type 1 vaccine in subjects at low risk of infection. J Infect Dis 168: 1387-1395.
- Wormser GP (1996) Lyme disease vaccine. Infection 24: 203-207.
- Doherty TM (2004) New vaccines against tuberculosis. TM & IH 9: 818-826.
- Reed SG, Bertholet S, Coler RN, Friede M (2009) New horizons in adjuvants for vaccine development. Trends Immunol 30: 23-32.
- Reed SG, Bertholet S, Coler RN, Friede M (2009) New horizons in adjuvants for vaccine development. Trends Immunol 30: 23-32.
- Kamath AT, Rochat AF, Valenti MP, Agger EM, Lingnau K, et al. (2008) Adult-like anti-mycobacterial T cell and in vivo dendritic cell responses following neonatal immunization with Ag85B-ESAT-6 in the IC31 adjuvant. PLoS One 3: e3683.
- Agger EM, Rosenkrands I, Hansen J, Brahimi K, Vandahl BS, et al. (2008) Cationic liposomes formulated with synthetic mycobacterial cordfactor (CAF01): a versatile adjuvant for vaccines with different immunological requirements. PLoS One 3: e3116.
- Kamath AT, Rochat AF, Christensen D, Agger EM, Andersen P, et al. (2009) A liposome-based mycobacterial vaccine induces potent adult and neonatal multifunctional T cells through the exquisite targeting of dendritic cells. PLoS One 4: e5771.
- Kamath AT, Valenti MP, Rochat AF, Agger EM, Lingnau K, et al. (2008) Protective anti-mycobacterial T cell responses through exquisite in vivo activation of vaccine-targeted dendritic cells. Eur J Immunol 38: 1247-1256.
- Skeiky YA, Alderson MR, Ovendale PJ, Guderian JA, Brandt L, et al. (2004) Differential immune responses and protective efficacy induced by components of a tuberculosis polyprotein vaccine, Mtb72F, delivered as naked DNA or recombinant protein. J Immunol 172: 7618-7628.
- Brandt L, Skeiky YA, Alderson MR, Lobet Y, Dalemans W, et al. (2004) The protective effect of the Mycobacterium bovis BCG vaccine is increased by coadministration with the Mycobacterium tuberculosis 72-kilodalton fusion polyprotein Mtb72F in M. tuberculosis-infected guinea pigs. Infect immun 72: 6622-6632.
- Reed S, Lobet Y (2005) Tuberculosis vaccine development; from mouse to man. Microbes and infection 7: 922-931.
- Von Eschen K, Morrison R, Braun M, Ofori-Anyinam O, De Kock E, et al. (2009) The candidate tuberculosis vaccine Mtb72F/AS02A: Tolerability and immunogenicity in humans. Hum vaccin 5: 475-482.
- Weinrich OA, van Pinxteren LA, Meng OL, Birk RP, Andersen P (2001) Protection of mice with a tuberculosis subunit vaccine based on a fusion protein of antigen 85b and ESAT-6. Infect Immun 69: 2773-2778.
- Leroux-Roels I, Leroux-Roels G, Ofori-Anyinam O, Moris P, De Kock E, et al. (2010) Evaluation of the safety and immunogenicity of two antigen concentrations of the Mtb72F/AS02(A) candidate tuberculosis vaccine in purified protein derivative-negative adults. Clin vaccine immunol : CVI 17: 1763-1771.
- Weinrich OA, van Pinxteren LA, Meng OL, Birk RP, Andersen P (2001) Protection of mice with a tuberculosis subunit vaccine based on a fusion protein of antigen 85b and ESAT-6. Infect Immun 69: 2773-2778.
- Andersen P, Munk ME, Pollock JM, Doherty TM (2000) Specific immune-based diagnosis of tuberculosis. Lancet 356: 1099-1104.
- Langermans JA, Doherty TM, Vervenne RA, van der Laan T, Lyashchenko K, et al. (2005) Protection of macaques against Mycobacterium tuberculosis infection by a subunit vaccine based on a fusion protein of antigen 85B and ESAT-6. Vaccine 23: 2740-2750.
- Agger EM, Rosenkrands I, Olsen AW, Hatch G, Williams A, et al. (2006) Protective immunity to tuberculosis with Ag85B-ESAT-6 in a synthetic cationic adjuvant system IC31. Vaccine 24: 5452-5460.
- Lindenstrom T, Agger EM, Korsholm KS, Darrah PA, Aagaard C, et al. (2009) Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J Immunol 182: 8047-8055.
- van Dissel JT, Arend SM, Prins C, Bang P, Tingskov PN, et al. (2010) Ag85B-ESAT-6 adjuvanted with IC31 promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in naive human volunteers. Vaccine 28: 3571-3581.
- van Dissel JT, Soonawala D, Joosten SA, Prins C, Arend SM, et al. (2011) Ag85B-ESAT-6 adjuvanted with IC31(R) promotes strong and long-lived Mycobacterium tuberculosis specific T cell responses in volunteers with previous BCG vaccination or tuberculosis infection. Vaccine 29: 2100-2109.
- Lindenstrom T, Agger EM, Korsholm KS, Darrah PA, Aagaard C, et al. (2009) Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J Immunol 182: 8047-8055.
- Skeiky YA, Dietrich J, Lasco TM, Stagliano K, Dheenadhayalan V, et al. (2010) Non-clinical efficacy and safety of HyVac4:IC31 vaccine administered in a BCG prime-boost regimen. Vaccine 28: 1084-1093.
- Hervas-Stubbs S, Majlessi L, Simsova M, Morova J, Rojas MJ, et al. (2006) High frequency of CD4+ T cells specific for the TB10.4 protein correlates with protection against Mycobacterium tuberculosis infection. Infect Immun 74: 3396-3407.
- Hawkridge T, Mahomed H (2011) Prospects for a new, safer and more effective TB vaccine. Paediatr Respir Rev 12: 46-51.
- Aagaard C, Hoang T, Dietrich J, Cardona PJ, Izzo A, et al. (2011) A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nature medicine 17: 189-194.
- Govender L, Abel B, Hughes EJ, Scriba TJ, Kagina BM, et al. (2010) Higher human CD4 T cell response to novel Mycobacterium tuberculosis latency associated antigens Rv2660 and Rv2659 in latent infection compared with tuberculosis disease. Vaccine 29: 51-57.
- https://www.aeras.org/portfolio/index.php
- Sun J, Deghmane AE, Soualhine H, Hong T, Bucci C, et al. (2007) Mycobacterium bovis BCG disrupts the interaction of Rab7 with RILP contributing to inhibition of phagosome maturation. J Leukoc Biol 82: 1437-1445.
- Bourgarit A, Carcelain G, Martinez V, Lascoux C, Delcey V, et al. (2006) Explosion of tuberculin-specific Th1-responses induces immune restoration syndrome in tuberculosis and HIV co-infected patients. AIDS 20: pF1-F7.
- Lienhardt C, Azzurri A, Amedei A, Fielding K, Sillah J, et al. (2002) Active tuberculosis in Africa is associated with reduced Th1 and increased Th2 activity in vivo. Eur J Immunol 32: 1605-1613.
- McDyer JF, Hackley MN, Walsh TE, Cook JL, Seder RA (1997) Patients with multidrug-resistant tuberculosis with low CD4+ T cell counts have impaired Th1 responses. J Immunol 158: 492-500.
- Grode L, Seiler P, Baumann S, Hess J, Brinkmann V, et al. (2005) Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guerin mutants that secrete listeriolysin. Clin Investig 115: 2472-2479.
- Portnoy DA, Chakraborty T, Goebel W, Cossart P (1992) Molecular determinants of Listeria monocytogenes pathogenesis. Infect Immun 60: 1263-1267.
- Honer zu Bentrup K, Russell DG (2001) Mycobacterial persistence: adaptation to a changing environment. Trends in microbiology 9: 597-605.
- Geoffroy C, Gaillard JL, Alouf JE, Berche P (1987) Purification, characterization, and toxicity of the sulfhydryl-activated hemolysin listeriolysin O from Listeria monocytogenes. Infect Immun 55: 1641-1646.
- Rosario M, Fulkerson J, Soneji S, Parker J, Im EJ, et al. (2010) Safety and immunogenicity of novel recombinant BCG and modified vaccinia virus Ankara vaccines in neonate rhesus macaques. Virol J 84: 7815-7821.
- Sun R, Skeiky YA, Izzo A, Dheenadhayalan V, Imam Z, et al. (2009) Novel recombinant BCG expressing perfringolysin O and the over-expression of key immunodominant antigens; pre-clinical characterization, safety and protection against challenge with Mycobacterium tuberculosis. Vaccine 27: 4412-4423.
- Schuck SD, Mueller H, Kunitz F, Neher A, Hoffmann H, et al. (2009) Identification of T-cell antigens specific for latent Mycobacterium tuberculosis infection. PloS one 4: e5590.
- Popoff MR, Bouvet P (2009) Clostridial toxins. Future microbiol 4: 1021-1064.
- Martin C, Williams A, Hernandez-Pando R, Cardona PJ, Gormley E, et al. (2006) The live Mycobacterium tuberculosis phoP mutant strain is more attenuated than BCG and confers protective immunity against tuberculosis in mice and guinea pigs. Vaccine 24: 3408-3419.
- Perez E, Samper S, Bordas Y, Guilhot C, Gicquel B, et al. (2001) An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol 41: 179-187.
- Frigui W, Bottai D, Majlessi L, Monot M, Josselin E, et al. (2008) Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS pathogens 4: e33.
- Frigui W, Bottai D, Majlessi L, Monot M, Josselin E, et al. (2008) Control of M. tuberculosis ESAT-6 secretion and specific T cell recognition by PhoP. PLoS pathogens 4: e33.
- Verreck FA, Vervenne RA, Kondova I, van Kralingen KW, Remarque EJ, et al. (2009) MVA.85A boosting of BCG and an attenuated, phoP deficient M. tuberculosis vaccine both show protective efficacy against tuberculosis in rhesus macaques. PloS one 4: e5264.
- Initiative TV. Promising TB Vaccine Candidate Progresses to Phase I Clinical Trials. May 25, 2011:[Available from: https://www.tbvi.eu/news-agenda/news/].
- Infante E, Aguilar LD, Gicquel B, Pando RH (2005) Immunogenicity and protective efficacy of the Mycobacterium tuberculosis fadD26 mutant. Clin Exp Immunol 141: 21-28.
- https://www.tbvi.eu/news-agenda/news/
- Ferrer NL, Gomez AB, Soto CY, Neyrolles O, Gicquel B, et al. (2009) Intracellular replication of attenuated Mycobacterium tuberculosis phoP mutant in the absence of host cell cytotoxicity. Microbes infect 11: 115-122.
- Cardona PJ, Asensio JG, Arbues A, Otal I, Lafoz C, et al. (2009) Extended safety studies of the attenuated live tuberculosis vaccine SO2 based on phoP mutant. Vaccine 27: 2499-2505.
- Andrews JR, Gandhi NR, Moodley P, Shah NS, Bohlken L, et al. (2008) Exogenous reinfection as a cause of multidrug-resistant and extensively drug-resistant tuberculosis in rural South Africa. J infect dis 198: 1582-1589.
- Sampson SL, Dascher CC, Sambandamurthy VK, Russell RG, Jacobs WR, Jr, et al. (2004) Protection elicited by a double leucine and pantothenate auxotroph of Mycobacterium tuberculosis in guinea pigs. Infection and immunity 72: 3031-3037.
- Sampson SL, Mansfield KG, Carville A, Magee DM, Quitugua T, et al. (2011) Extended safety and efficacy studies of a live attenuated double leucine and pantothenate auxotroph of Mycobacterium tuberculosis as a vaccine candidate. Vaccine 29: 4839-4847.
- Vilaplana C, Montane E, Pinto S, Barriocanal AM, Domenech G, et al. (2010) Double-blind, randomized, placebo-controlled Phase I Clinical Trial of the therapeutical antituberculous vaccine RUTI. Vaccine 28: 1106-1116.
- Cardona PJ, Amat I, Gordillo S, Arcos V, Guirado E, et al. (2005) Immunotherapy with fragmented Mycobacterium tuberculosis cells increases the effectiveness of chemotherapy against a chronical infection in a murine model of tuberculosis. Vaccine 23: 1393-1398.
- Guirado E, Gil O, Caceres N, Singh M, Vilaplana C, et al. (2008) Induction of a specific strong polyantigenic cellular immune response after short-term chemotherapy controls bacillary reactivation in murine and guinea pig experimental models of tuberculosis. Clin vaccine immunol : CVI 15: 1229-1237.
- Vilaplana C, Gil O, Caceres N, Pinto S, Diaz J, et al. (2011) Prophylactic Effect of a Therapeutic Vaccine against TB Based on Fragments of Mycobacterium tuberculosis. PloS one 6: e20404.
- Faludi I, Szabo AM, Burian K, Endresz V, Miczak A (2011) Recombinant Mycobacterium smegmatis vaccine candidates. Acta Microbiol Immunol Hung 58: 13-22.
- Lindsey DR, Dhandayuthapani S, Jagannath C (2009) Anti-tuberculosis immunity induced in mice by vaccination with Mycobacterium smegmatis over-expressing Antigen 85B is due to the increased influx of IFNgamma-positive CD4 T cells into the lungs. Tuberculosis (Edinb) 89 Suppl 1: S46-48.
- Zhang H, Peng P, Miao S, Zhao Y, Mao F, et al. (2010) Recombinant Mycobacterium smegmatis expressing an ESAT6-CFP10 fusion protein induces anti-mycobacterial immune responses and protects against Mycobacterium tuberculosis challenge in mice. Scand J Immunol 72: 349-357.
- Yi Z, Fu Y, Yang C, Li J, Luo X, et al. (2007) Recombinant M. smegmatis vaccine targeted delivering IL-12/GLS into macrophages can induce specific cellular immunity against M. tuberculosis in BALB/c mice. Vaccine 25: 638-648.
- Lahey T, Arbeit RD, Bakari M, Horsburgh CR, Matee M, et al. (2010) Immunogenicity of a protective whole cell mycobacterial vaccine in HIV-infected adults: a phase III study in Tanzania. Vaccine 28: 7652-7658.
- Goldwater PN (2006) A pilot study of SRL 172 (killed Mycobacterium vaccae) in healthy chronic hepatitis B carriers and hepatitis B vaccine non-responders. Hum Vaccin 2: 8-13.
- O'Brien ME, Saini A, Smith IE, Webb A, Gregory K, et al. (2000) A randomized phase II study of SRL172 (Mycobacterium vaccae) combined with chemotherapy in patients with advanced inoperable non-small-cell lung cancer and mesothelioma. Br J Cancer 83: 853-857.
- Waddell RD, Chintu C, Lein AD, Zumla A, Karagas MR, et al. (2000) Safety and immunogenicity of a five-dose series of inactivated Mycobacterium vaccae vaccination for the prevention of HIV-associated tuberculosis. Clin Infect Dis 30 Suppl 3: S309-15.
- von Reyn CF, Arbeit RD, Yeaman G, Waddell RD, Marsh BJ, et al. (1997) Immunization of healthy adult subjects in the United States with inactivated Mycobacterium vaccae administered in a three-dose series. Clin Infect Dis 24: 843-848.
- Stanford JL, Stanford CA (1994) Immunotherapy of tuberculosis with Mycobacterium vaccae NCTC 11659. Immunobiology 191: 555-563.
- Stanford JL, Rook GA, Bahr GM, Dowlati Y, Ganapati R, et al. (1990) Mycobacterium vaccae in immunoprophylaxis and immunotherapy of leprosy and tuberculosis. Vaccine 8: 525-530.
- von Reyn CF, Mtei L, Arbeit RD, Waddell R, Cole B, et al. (2010) Prevention of tuberculosis in Bacille Calmette-Guerin-primed, HIV-infected adults boosted with an inactivated whole-cell mycobacterial vaccine. AIDS 24: 675-685.
- Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, et al. (2009) Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med 15: 293-299.
- Sung H, Schleiss MR (2010) Update on the current status of cytomegalovirus vaccines. Expert Rev Vaccines 9: 1303-1314.
- Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, et al. (2011) Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473: 523-527.
- Pethe K, Alonso S, Biet F, Delogu G, Brennan MJ, et al. (2001) The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature 412: 190-194.
- Locht C, Hougardy JM, Rouanet C, Place S, Mascart F (2006) Heparin-binding hemagglutinin, from an extrapulmonary dissemination factor to a powerful diagnostic and protective antigen against tuberculosis. Tuberculosis (Edinb) 86: 303-309.
- Guerrero GG, Locht C (2011) Recombinant HBHA boosting effect on BCG-induced immunity against Mycobacterium tuberculosis infection. Clin Dev Immunol 2011: 730702.
- Parra M, Pickett T, Delogu G, Dheenadhayalan V, Debrie AS, et al. (2004) The mycobacterial heparin-binding hemagglutinin is a protective antigen in the mouse aerosol challenge model of tuberculosis. Infect Immun 72: 6799-6805.
- Temmerman S, Pethe K, Parra M, Alonso S, Rouanet C, et al. (2004) Methylation-dependent T cell immunity to Mycobacterium tuberculosis heparin-binding hemagglutinin. Nat Med 10: 935-941.
- Okada M, Kita Y (2010) Tuberculosis vaccine development: The development of novel (preclinical) DNA vaccine. Hum Vaccin 6: 297-308.
- Nor NM, Musa M (2004) Approaches towards the development of a vaccine against tuberculosis: recombinant BCG and DNA vaccine. Tuberculosis (Edinb) 84: 102-109.
- Taylor JJ, Jenkins MK (2011) CD4+ memory T cell survival. Curr opin immunol 23: 319-323.
- Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM (2006) Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity 24: 677-688.
- Puzo G (1990) The carbohydrate- and lipid-containing cell wall of mycobacteria, phenolic glycolipids: structure and immunological properties. Crit rev microbiol 17: 305-327.
Relevant Topics
- Anthrax Bioterrorism
- Bio surveilliance
- Biodefense
- Biohazards
- Biological Preparedness
- Biological Warfare
- Biological weapons
- Biorisk
- Bioterrorism
- Bioterrorism Agents
- Biothreat Agents
- Disease surveillance
- Emerging infectious disease
- Epidemiology of Breast Cancer
- Information Security
- Mass Prophylaxis
- Nuclear Terrorism
- Probabilistic risk assessment
- United States biological defense program
- Vaccines
Recommended Journals
Article Tools
Article Usage
- Total views: 15973
- [From(publication date):
specialissue-2012 - Dec 19, 2025] - Breakdown by view type
- HTML page views : 11079
- PDF downloads : 4894