 Spanish
Spanish  Chinese
Chinese  Russian
Russian  German
German  French
French  Japanese
Japanese  Portuguese
Portuguese  Hindi
Hindi Our Group organises 3000+ Global Conferenceseries Events every year across USA, Europe & Asia with support from 1000 more scientific Societies and Publishes 700+ Open Access Journals which contains over 50000 eminent personalities, reputed scientists as editorial board members.
Open Access Journals gaining more Readers and Citations
700 Journals and 15,000,000 Readers Each Journal is getting 25,000+ Readers
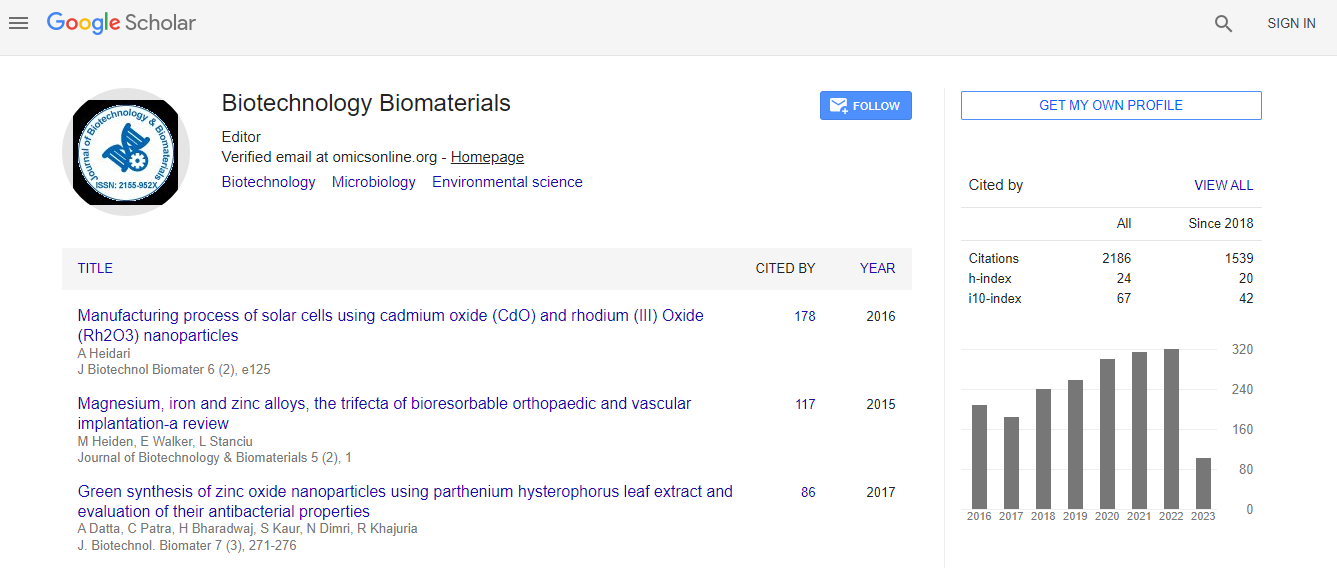
Google Scholar citation report
Citations : 3330
Journal of Biotechnology & Biomaterials received 3330 citations as per Google Scholar report
Indexed In
- Index Copernicus
- Google Scholar
- Sherpa Romeo
- Open J Gate
- Genamics JournalSeek
- Academic Keys
- ResearchBible
- China National Knowledge Infrastructure (CNKI)
- Access to Global Online Research in Agriculture (AGORA)
- Electronic Journals Library
- RefSeek
- Hamdard University
- EBSCO A-Z
- OCLC- WorldCat
- SWB online catalog
- Virtual Library of Biology (vifabio)
- Publons
- Geneva Foundation for Medical Education and Research
- Euro Pub
- ICMJE
Useful Links
Recommended Journals
Related Subjects
Share This Page
In Association with
Silencing the mutant HuntingtonГўВ?В?s disease gene using CRISPR-Cas9
18th Biotechnology Congress
N Kolli, M Lu, P Maiti, J Rossignol and G L Dunbar
Central Michigan University, USA St. Mary's of Michigan - Field Neurosciences Institute, USA
ScientificTracks Abstracts: J Biotechnol Biomater
Abstract
HuntingtonГўВ?В?s disease (HD) is a hereditary, fatal neurodegenerative disorder that is most prominently characterized by death of striatal neurons, chorea, as well as cognitive and emotional dysfunction. The mutant HuntingtonГўВ?В?s gene (mHTT) contains extra ГўВ?В?CAGГўВ?В? codon repeats in the exon1 from which the translated huntingtin protein (HTT) gains an elongated glutamine tract that prevents the protein from undergoing normal post-translational modifications. In addition to its non-functional property, this mutant protein confers a toxic gain of function, which is responsible for the pathophysiology of the HD. Although several studies have shown behavioral sparing in the animal models of HD through the use of allogeneic and autologous stem-cell transplantation, the existence of the mutant HTT prevents long-term functional recovery. For this reason, various gene silencing techniques, such as RNA interference mechanisms, have been extensively studied during the past decade. However, these RNA interference strategies are less efficient and require multiple treatments at approximately six-month intervals. As a promising alternative approach, we propose that the most efficient way to treat HD may be to curb the production of the mutant HTT itself, through the use of the gene editing tool, the CRISPR-Cas9 system. We have constructed two CRISPR (clustered regularly interspaced short palindromic repeat)Cas9 (CRISPR associate protein) plasmids, among which one nicks the DNA at untranslated region upstream to the open reading frame (uORF), and the other nicks the DNA at exon1-intron boundary. The primary goal of this study was to apply this plasmid into mesenchymal stem cells (MSCs) extracted from the bone-marrow of YAC128 mice, which carries the transgene for HD. Our results suggested that the disruption of uORF through CRISPR-Cas9 influences the translation of mHTT negatively and, to a lesser extent, disrupts the exon1-intron boundary, which affects the translation of the mHTT. These findings also revealed the pattern of the nucleotide addition or deletion at the site of the DNA-nick in this modelBiography
Nivya Kolli is a doctoral candidate in the program of Neuroscience at Central Michigan University, USA. She received her Doctor of Pharmacy degree from Manipal University, India and has multiple research experiences, both, at national and international level. Her research seeks to understand molecular and cellular aspects related to the pathology associated with mutant Huntingtin protein. She is currently investigating the potential of CRISPR-Cas9 mediated gene-silencing and gene-correction to treat Huntington’s disease. She also work with the team at Field Neurosciences Institute, Michigan, to investigate the downstream autophagic pathway involved in traumatic brain injury and the use of curcumin as a potential therapeutic treatment for Alzheimer’s disease and glioblastoma. Her expertise is in studying gene expression, gene-editing, toxicology, cell pharmacology and pharmacokinetics.